Department of Theoretical Biophysics, Max Planck Institute of Biophysics, Frankfurt am Main, Germany.
Faculty of Physics, University of Vienna, Vienna, Austria.
PLoS Comput Biol. 2021 Apr 1;17(4):e1008790. doi: 10.1371/journal.pcbi.1008790. eCollection 2021 Apr.
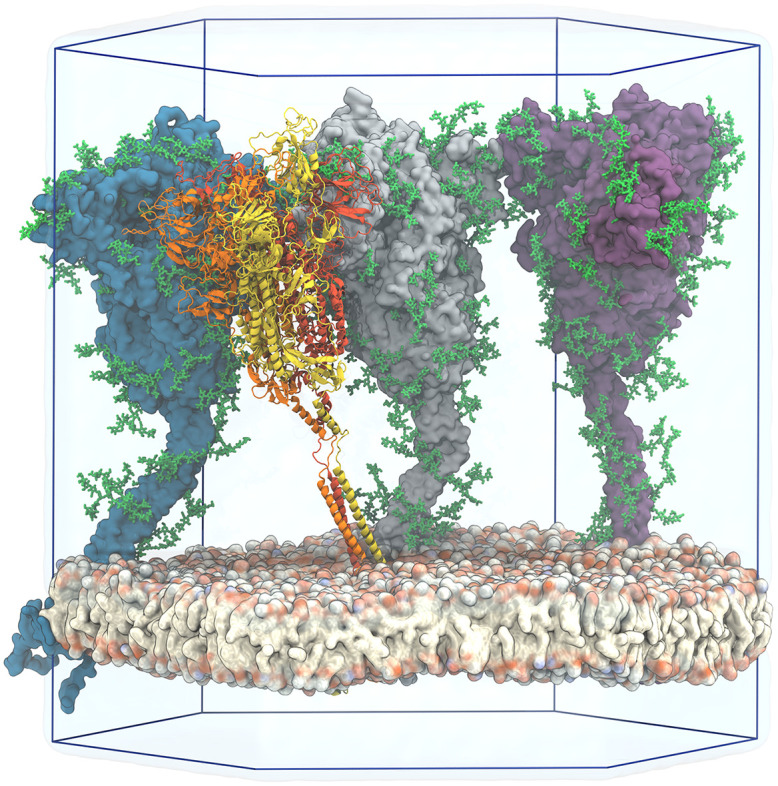
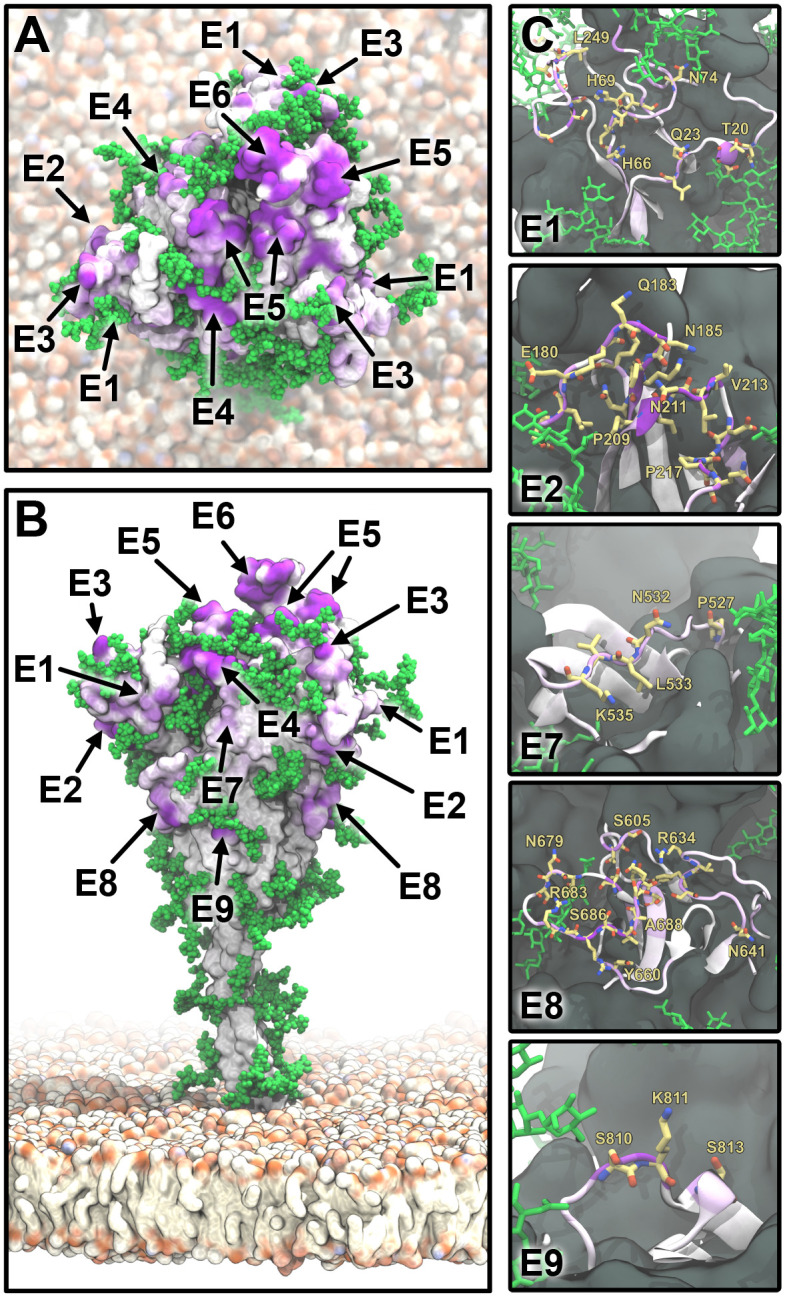
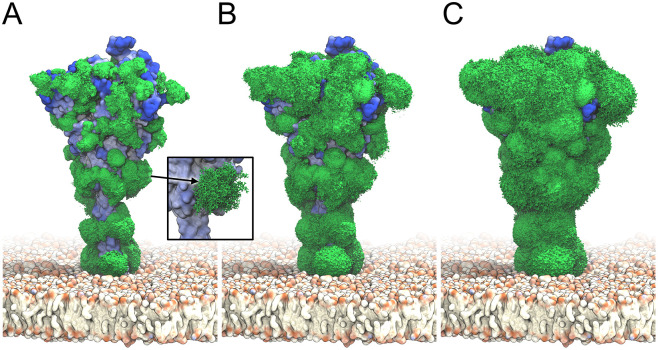
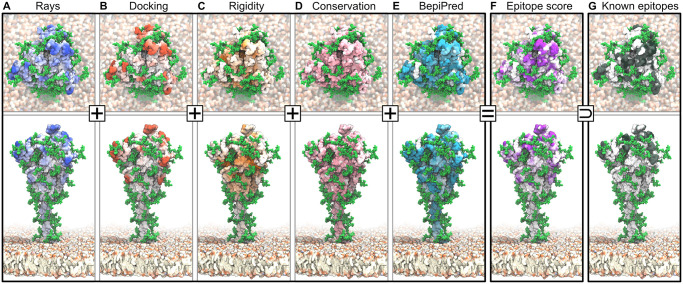
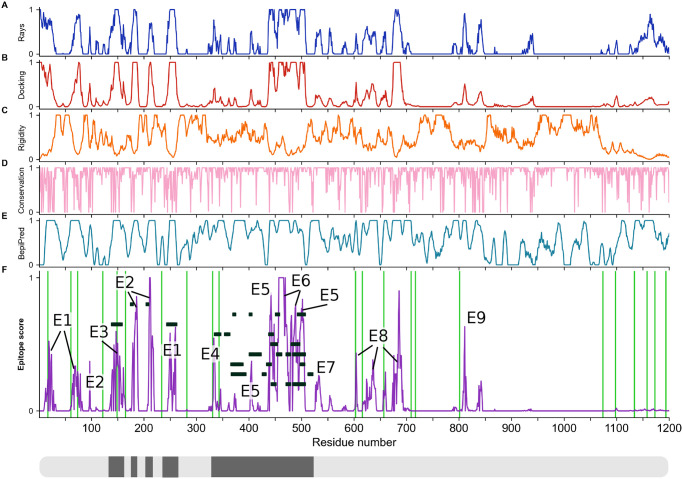
The primary immunological target of COVID-19 vaccines is the SARS-CoV-2 spike (S) protein. S is exposed on the viral surface and mediates viral entry into the host cell. To identify possible antibody binding sites, we performed multi-microsecond molecular dynamics simulations of a 4.1 million atom system containing a patch of viral membrane with four full-length, fully glycosylated and palmitoylated S proteins. By mapping steric accessibility, structural rigidity, sequence conservation, and generic antibody binding signatures, we recover known epitopes on S and reveal promising epitope candidates for structure-based vaccine design. We find that the extensive and inherently flexible glycan coat shields a surface area larger than expected from static structures, highlighting the importance of structural dynamics. The protective glycan shield and the high flexibility of its hinges give the stalk overall low epitope scores. Our computational epitope-mapping procedure is general and should thus prove useful for other viral envelope proteins whose structures have been characterized.
COVID-19 疫苗的主要免疫目标是 SARS-CoV-2 刺突(S)蛋白。S 蛋白暴露在病毒表面,介导病毒进入宿主细胞。为了识别可能的抗体结合位点,我们对包含带有四个全长、完全糖基化和棕榈酰化 S 蛋白的病毒膜斑块的 410 万个原子系统进行了多微秒分子动力学模拟。通过映射空间可达性、结构刚性、序列保守性和通用抗体结合特征,我们恢复了 S 蛋白上的已知表位,并揭示了基于结构的疫苗设计有希望的表位候选物。我们发现广泛而固有的灵活聚糖涂层覆盖了比静态结构预期更大的表面积,突出了结构动力学的重要性。保护性聚糖屏蔽和其铰链的高灵活性使茎总体上具有较低的表位评分。我们的计算表位作图程序是通用的,因此应该对其他结构特征已被表征的病毒包膜蛋白有用。