Program in Bioinformatics & Computational Biology, Iowa State University, Ames, Iowa, United States of America.
Department of Plant Pathology & Microbiology, Iowa State University, Ames, Iowa, United States of America.
PLoS Comput Biol. 2021 Apr 2;17(4):e1008890. doi: 10.1371/journal.pcbi.1008890. eCollection 2021 Apr.
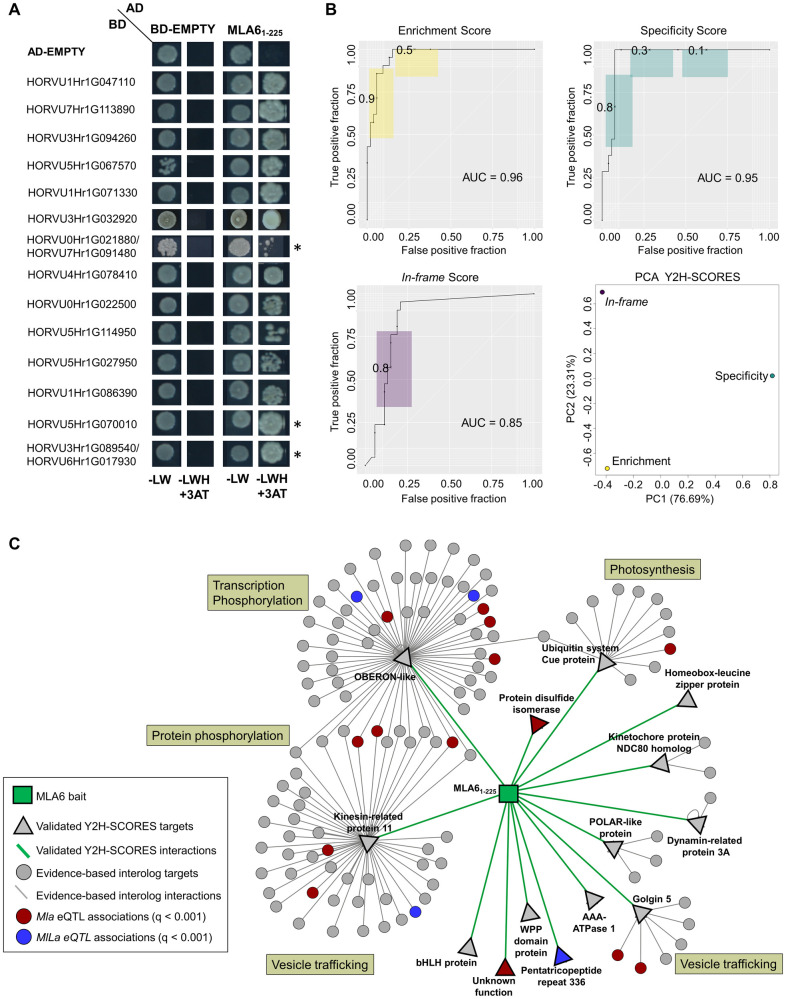
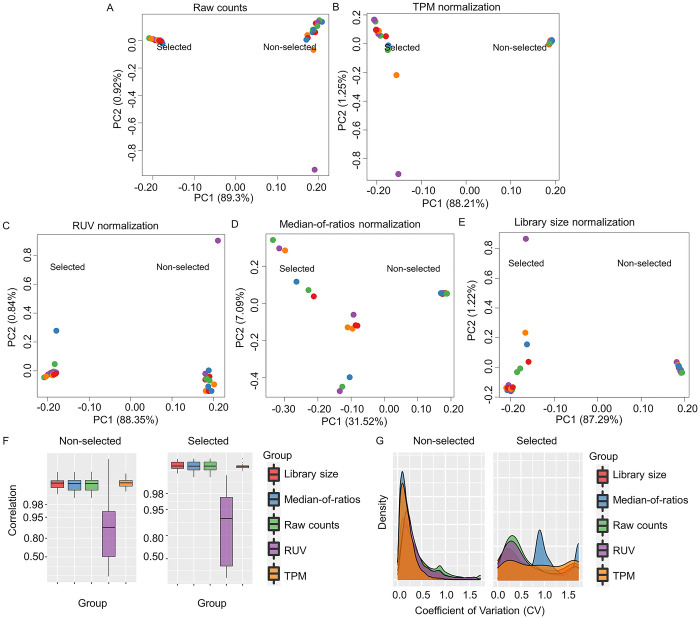
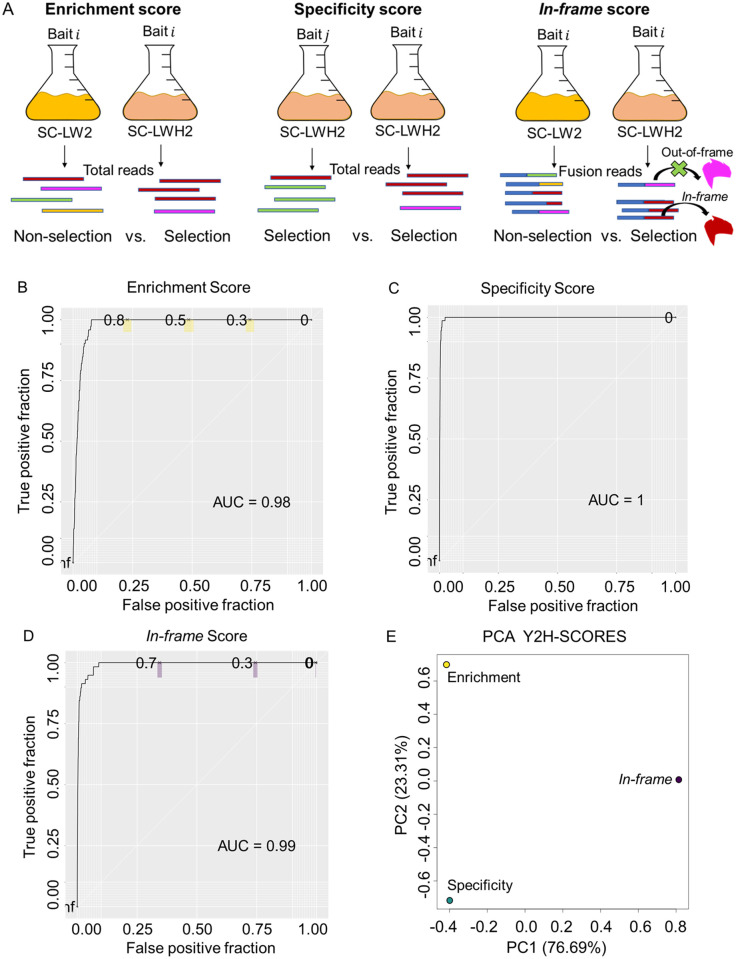
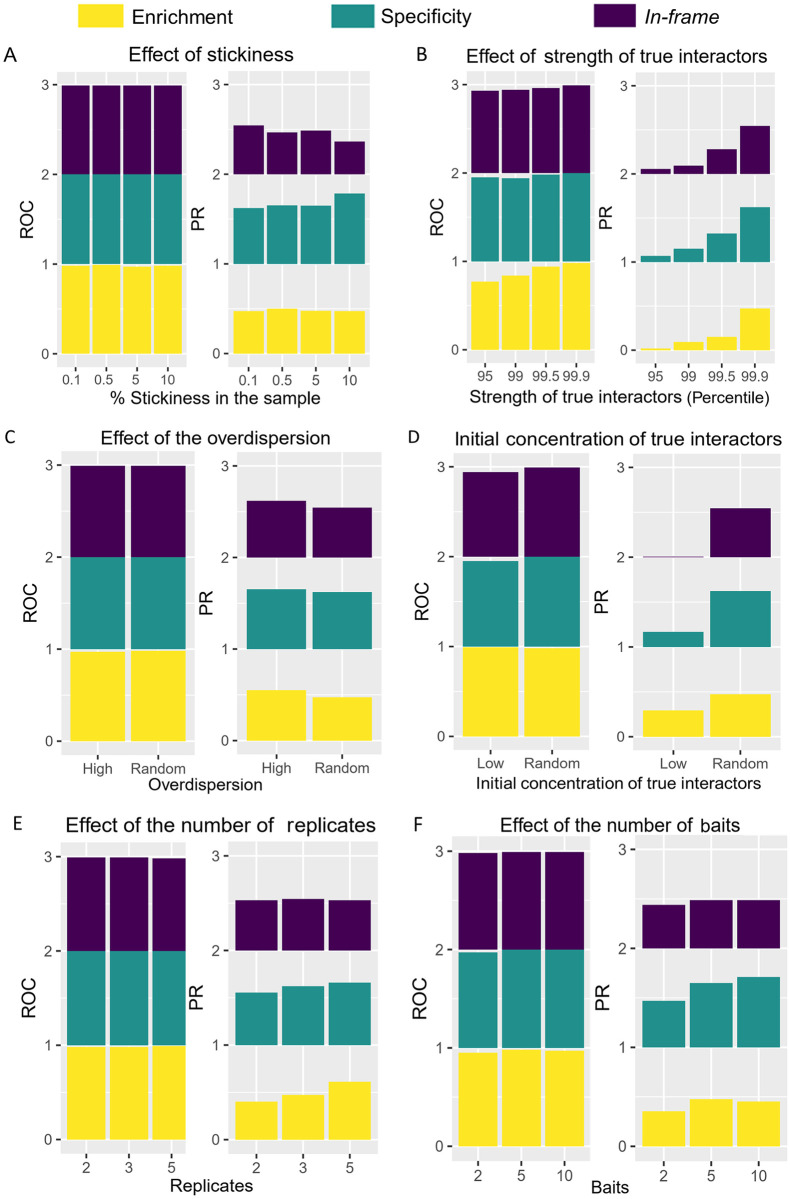
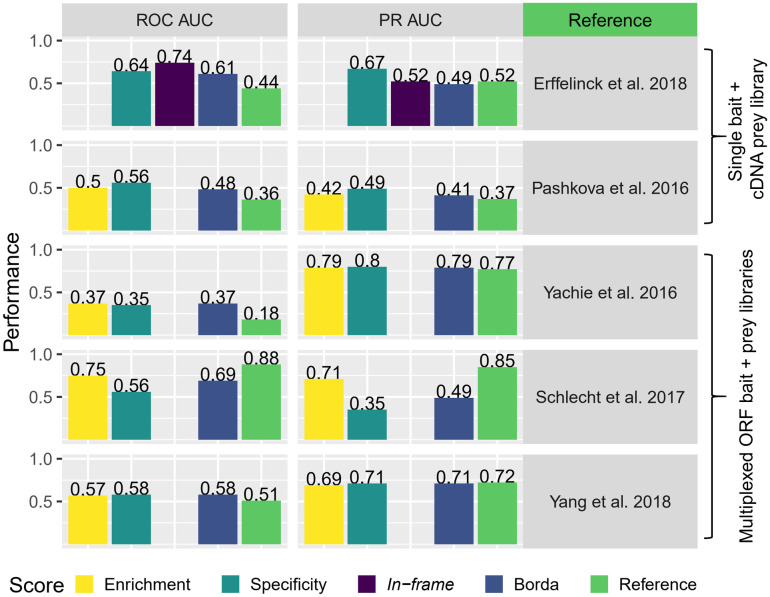
Protein-protein interaction networks are one of the most effective representations of cellular behavior. In order to build these models, high-throughput techniques are required. Next-generation interaction screening (NGIS) protocols that combine yeast two-hybrid (Y2H) with deep sequencing are promising approaches to generate interactome networks in any organism. However, challenges remain to mining reliable information from these screens and thus, limit its broader implementation. Here, we present a computational framework, designated Y2H-SCORES, for analyzing high-throughput Y2H screens. Y2H-SCORES considers key aspects of NGIS experimental design and important characteristics of the resulting data that distinguish it from RNA-seq expression datasets. Three quantitative ranking scores were implemented to identify interacting partners, comprising: 1) significant enrichment under selection for positive interactions, 2) degree of interaction specificity among multi-bait comparisons, and 3) selection of in-frame interactors. Using simulation and an empirical dataset, we provide a quantitative assessment to predict interacting partners under a wide range of experimental scenarios, facilitating independent confirmation by one-to-one bait-prey tests. Simulation of Y2H-NGIS enabled us to identify conditions that maximize detection of true interactors, which can be achieved with protocols such as prey library normalization, maintenance of larger culture volumes and replication of experimental treatments. Y2H-SCORES can be implemented in different yeast-based interaction screenings, with an equivalent or superior performance than existing methods. Proof-of-concept was demonstrated by discovery and validation of novel interactions between the barley nucleotide-binding leucine-rich repeat (NLR) immune receptor MLA6, and fourteen proteins, including those that function in signaling, transcriptional regulation, and intracellular trafficking.
蛋白质-蛋白质相互作用网络是细胞行为最有效的表示之一。为了构建这些模型,需要高通量技术。将酵母双杂交 (Y2H) 与深度测序相结合的下一代相互作用筛选 (NGIS) 协议是在任何生物体中生成相互作用网络的有前途的方法。然而,从这些筛选中挖掘可靠信息仍然存在挑战,因此限制了其更广泛的实施。在这里,我们提出了一种计算框架,指定为 Y2H-SCORES,用于分析高通量 Y2H 筛选。Y2H-SCORES 考虑了 NGIS 实验设计的关键方面和区分它与 RNA-seq 表达数据集的重要数据特征。实施了三个定量排名分数来识别相互作用伙伴,包括:1)对阳性相互作用的选择有显著富集,2)多诱饵比较之间的相互作用特异性程度,以及 3)选择框内相互作用体。使用模拟和经验数据集,我们提供了一种定量评估,以在广泛的实验场景下预测相互作用伙伴,通过一对一诱饵-猎物测试进行独立确认。Y2H-NGIS 的模拟使我们能够确定最大化检测真实相互作用体的条件,这些条件可以通过诱饵文库归一化、保持更大的培养体积和复制实验处理等协议来实现。Y2H-SCORES 可以在不同的基于酵母的相互作用筛选中实施,具有与现有方法相当或更好的性能。通过发现和验证大麦核苷酸结合亮氨酸重复 (NLR) 免疫受体 MLA6 与十四个蛋白质之间的新相互作用,证明了概念验证,包括那些在信号转导、转录调节和细胞内运输中起作用的蛋白质。