Pértille Fabio, Alvarez-Rodriguez Manuel, da Silva Arthur Nery, Barranco Isabel, Roca Jordi, Guerrero-Bosagna Carlos, Rodriguez-Martinez Heriberto
Department of Physics, Chemistry and Biology, Linköping University, SE-58183 Linköping, Sweden.
Department of Biomedical & Clinical Sciences (BKV), Linköping University, SE-58185 Linköping, Sweden.
Int J Mol Sci. 2021 Mar 6;22(5):2679. doi: 10.3390/ijms22052679.
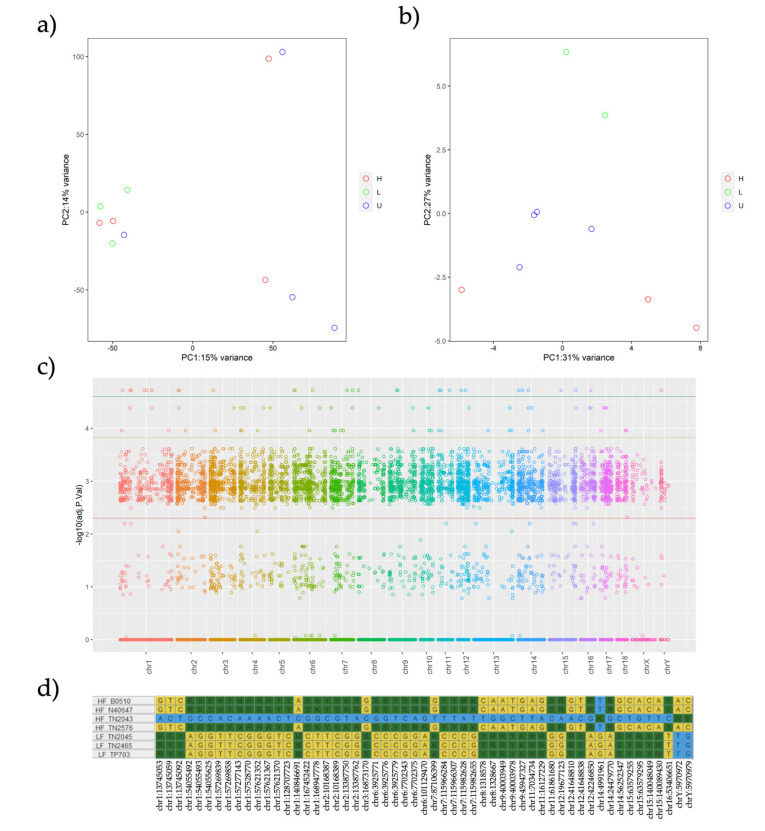
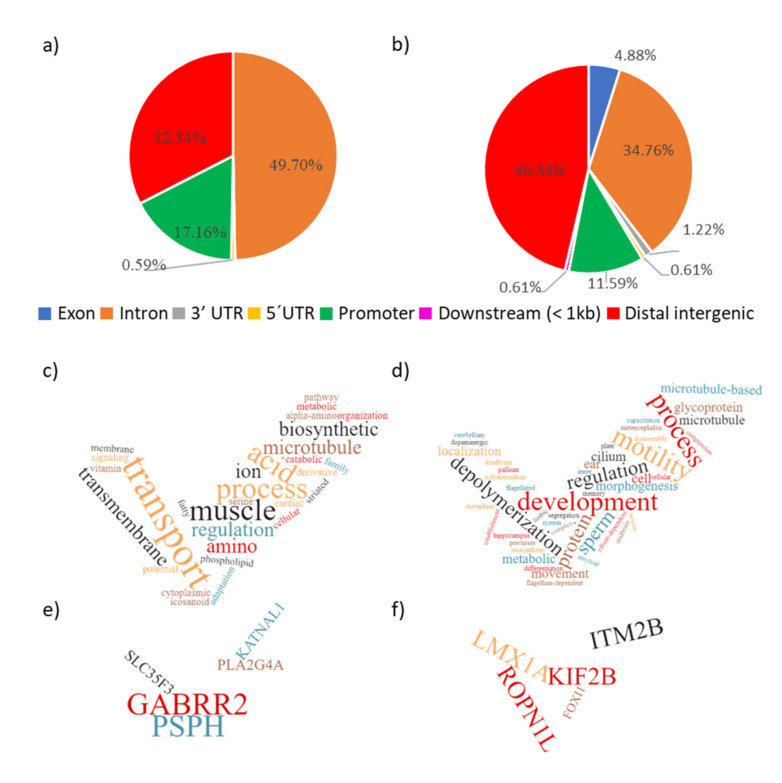
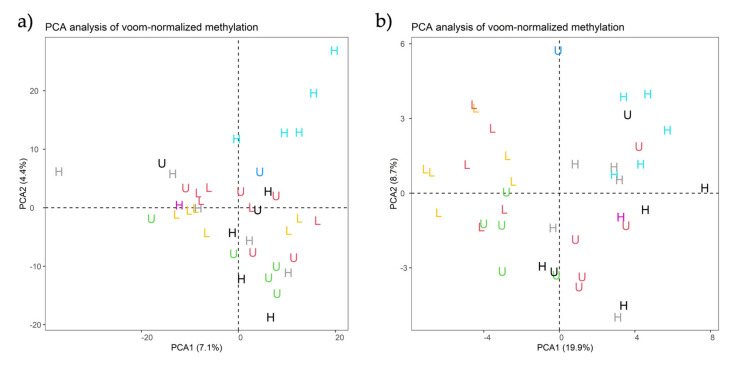
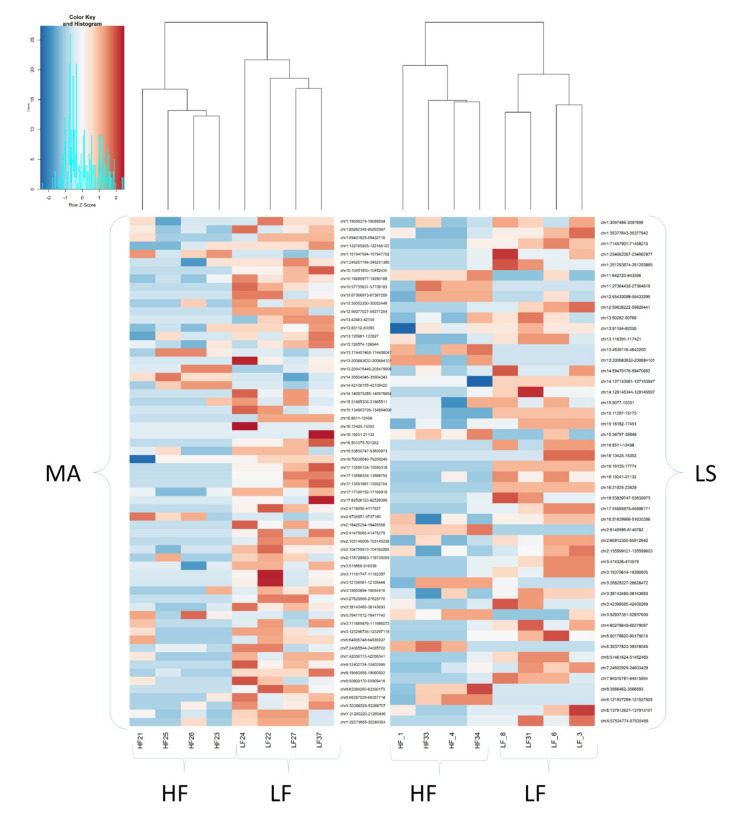
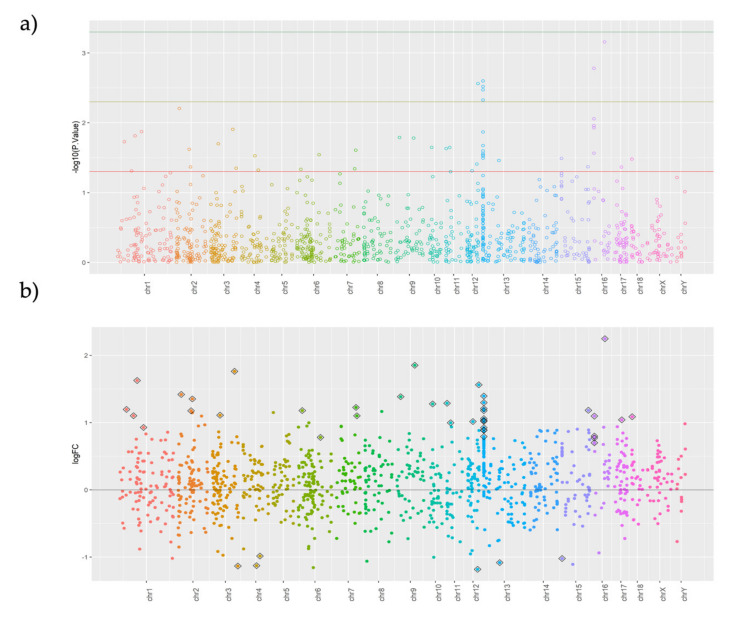
A combined Genotyping By Sequencing (GBS) and methylated DNA immunoprecipitation (MeDIP) protocol was used to identify-in parallel-genetic variation (Genomic-Wide Association Studies (GWAS) and epigenetic differences of Differentially Methylated Regions (DMR) in the genome of spermatozoa from the porcine animal model. Breeding boars with good semen quality ( = 11) and specific and well-documented differences in fertility (farrowing rate, FR) and prolificacy (litter size, LS) ( = 7) in artificial insemination programs, using combined FR and LS, were categorized as High Fertile (HF, = 4) or Low Fertile (LF, = 3), and boars with Unknown Fertility (UF, = 4) were tested for eventual epigenetical similarity with those fertility-proven. We identified 165,944 Single Nucleotide Polymorphisms (SNPs) that explained 14-15% of variance among selection lines. Between HF and LF individuals ( = 7, 4 HF and 3 LF), we identified 169 SNPs with ≤ 0.00015, which explained 58% of the variance. For the epigenetic analyses, we considered fertility and period of ejaculate collection (late-summer and mid-autumn). Approximately three times more DMRs were observed in HF than in LF boars across these periods. Interestingly, UF boars were clearly clustered with one of the other HF or LF groups. The highest differences in DMRs between HF and LF experimental groups across the pig genome were located in the chr 3, 9, 13, and 16, with most DMRs being hypermethylated in LF boars. In both HF and LF boars, DMRs were mostly hypermethylated in late-summer compared to mid-autumn. Three overlaps were detected between SNPs ( ≤ 0.0005, = 1318) and CpG sites within DMRs. In conclusion, fertility levels in breeding males including FR and LS can be discerned using methylome analyses. The findings in this biomedical animal model ought to be applied besides sire selection for andrological diagnosis of idiopathic sub/infertility.
采用一种结合测序基因分型(GBS)和甲基化DNA免疫沉淀(MeDIP)的方案,以并行方式鉴定猪动物模型精子基因组中的遗传变异(全基因组关联研究,GWAS)和差异甲基化区域(DMR)的表观遗传差异。在人工授精项目中,根据繁殖公猪的精液质量(n = 11)以及生育力(产仔率,FR)和繁殖力(窝产仔数,LS)方面明确记录的特定差异(n = 7),结合FR和LS,将公猪分为高生育力(HF,n = 4)或低生育力(LF,n = 3),并对生育力未知(UF,n = 4)的公猪与已证实生育力的公猪进行表观遗传相似性测试。我们鉴定出165,944个单核苷酸多态性(SNP),这些SNP解释了选择品系间14 - 15%的变异。在HF和LF个体(n = 7,4头HF和3头LF)之间,我们鉴定出169个P值≤0.00015的SNP,这些SNP解释了58%的变异。对于表观遗传分析,我们考虑了生育力和射精采集时期(夏末和中秋)。在这些时期,HF公猪中观察到的DMR数量大约是LF公猪的三倍。有趣的是,UF公猪明显聚集在其他HF或LF组中的一组。猪基因组中HF和LF实验组之间DMR差异最大的区域位于3号、9号、13号和16号染色体,大多数DMR在LF公猪中呈高甲基化。在HF和LF公猪中,与中秋相比,夏末时DMR大多呈高甲基化。在DMR内的SNP(P≤0.0005,n = 1318)和CpG位点之间检测到三个重叠。总之,使用甲基化组分析可以辨别包括FR和LS在内的种公猪的生育力水平。除了用于雄性生殖疾病特发性亚不育/不育的父本选择和男科诊断外,该生物医学动物模型的研究结果也应得到应用。