Department of Cardiology, St. Antonius Hospital, 3435 CM Nieuwegein, The Netherlands.
Department of Pulmonology, St. Antonius Hospital, 3435 CM Nieuwegein, The Netherlands.
Int J Mol Sci. 2021 Mar 27;22(7):3471. doi: 10.3390/ijms22073471.
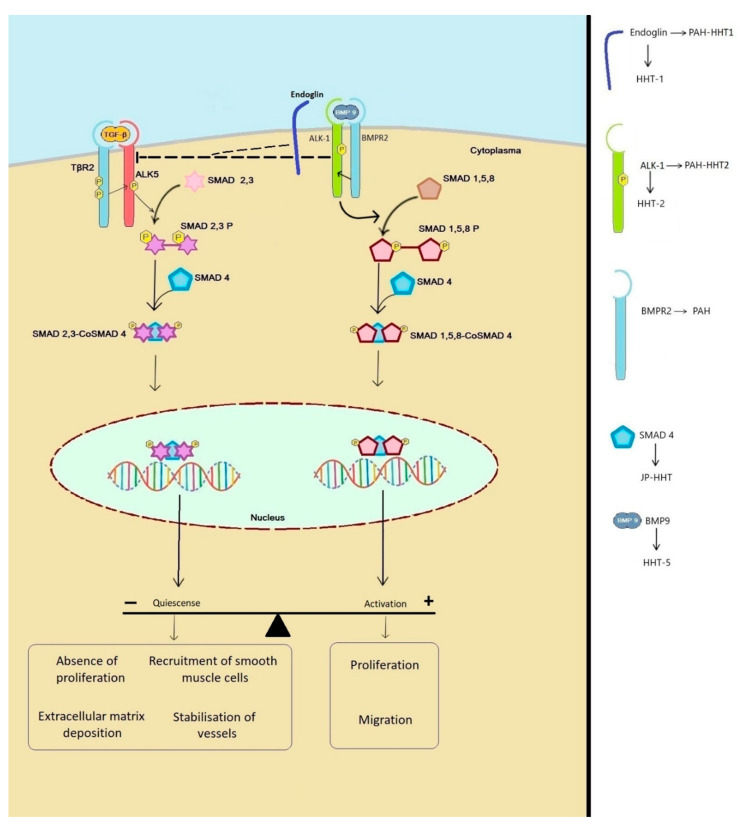
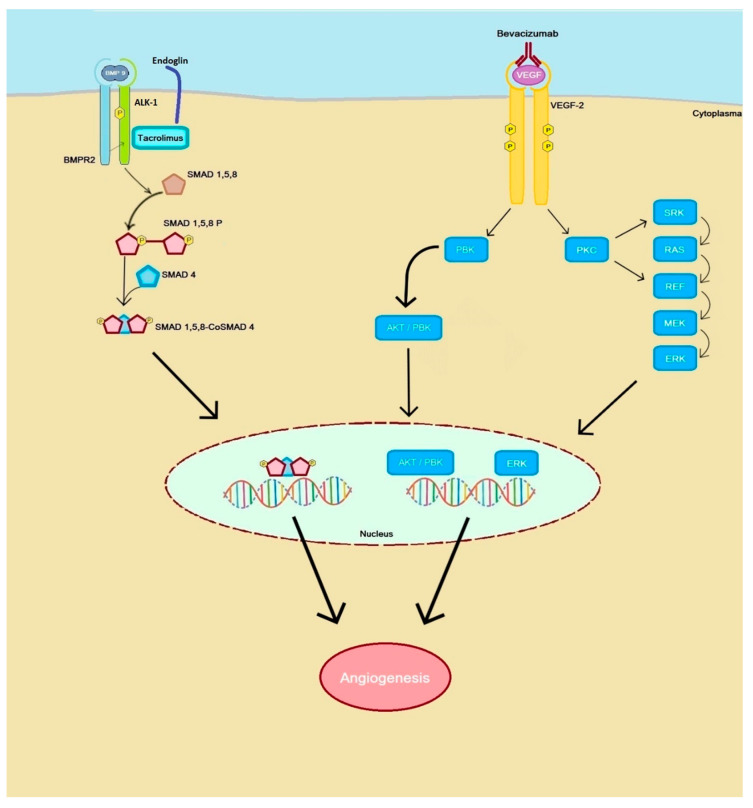
In this review, we discuss the role of transforming growth factor-beta (TGF-β) in the development of pulmonary vascular disease (PVD), both pulmonary arteriovenous malformations (AVM) and pulmonary hypertension (PH), in hereditary hemorrhagic telangiectasia (HHT). HHT or Rendu-Osler-Weber disease is an autosomal dominant genetic disorder with an estimated prevalence of 1 in 5000 persons and characterized by epistaxis, telangiectasia and AVMs in more than 80% of cases, HHT is caused by a mutation in the ENG gene on chromosome 9 encoding for the protein endoglin or activin receptor-like kinase 1 (ACVRL1) gene on chromosome 12 encoding for the protein ALK-1, resulting in HHT type 1 or HHT type 2, respectively. A third disease-causing mutation has been found in the SMAD-4 gene, causing a combination of HHT and juvenile polyposis coli. All three genes play a role in the TGF-β signaling pathway that is essential in angiogenesis where it plays a pivotal role in neoangiogenesis, vessel maturation and stabilization. PH is characterized by elevated mean pulmonary arterial pressure caused by a variety of different underlying pathologies. HHT carries an additional increased risk of PH because of high cardiac output as a result of anemia and shunting through hepatic AVMs, or development of pulmonary arterial hypertension due to interference of the TGF-β pathway. HHT in combination with PH is associated with a worse prognosis due to right-sided cardiac failure. The treatment of PVD in HHT includes medical or interventional therapy.
在这篇综述中,我们讨论了转化生长因子-β(TGF-β)在遗传性出血性毛细血管扩张症(HHT)中肺血管疾病(PVD)的发展中的作用,包括肺动静脉畸形(AVM)和肺动脉高压(PH)。HHT 或朗道-奥尔斯勒-韦伯病是一种常染色体显性遗传疾病,估计患病率为每 5000 人中有 1 人,其特征是 80%以上的病例鼻出血、毛细血管扩张和 AVM。HHT 是由 ENG 基因(位于 9 号染色体上)或编码蛋白激活素受体样激酶 1(ALK-1)的 ACVRL1 基因(位于 12 号染色体上)的突变引起的,ENG 基因编码的蛋白为内皮糖蛋白,ACVRL1 基因编码的蛋白为 ALK-1,分别导致 HHT 1 型或 HHT 2 型。第三种致病突变已在 SMAD-4 基因中发现,导致 HHT 和青少年结肠息肉病的组合。这三个基因都在 TGF-β 信号通路中发挥作用,该通路对血管生成至关重要,在血管生成中,它在新血管生成、血管成熟和稳定中发挥关键作用。PH 的特征是平均肺动脉压升高,由多种不同的潜在病理引起。由于贫血和通过肝 AVM 的分流导致的高心输出量,或由于 TGF-β 途径的干扰导致的肺动脉高压的发展,HHT 增加了 PH 的风险。由于右侧心力衰竭,HHT 合并 PH 的预后较差。HHT 中 PVD 的治疗包括药物或介入治疗。