Aguilar-Ordoñez Israel, Pérez-Villatoro Fernando, García-Ortiz Humberto, Barajas-Olmos Francisco, Ballesteros-Villascán Judith, González-Buenfil Ram, Fresno Cristobal, Garcíarrubio Alejandro, Fernández-López Juan Carlos, Tovar Hugo, Hernández-Lemus Enrique, Orozco Lorena, Soberón Xavier, Morett Enrique
Instituto de Biotecnología, Universidad Nacional Autónoma de México (UNAM), Cuernavaca, Morelos, México.
Instituto Nacional de Medicina Genómica (INMEGEN), Mexico City, México.
PLoS One. 2021 Apr 8;16(4):e0249773. doi: 10.1371/journal.pone.0249773. eCollection 2021.
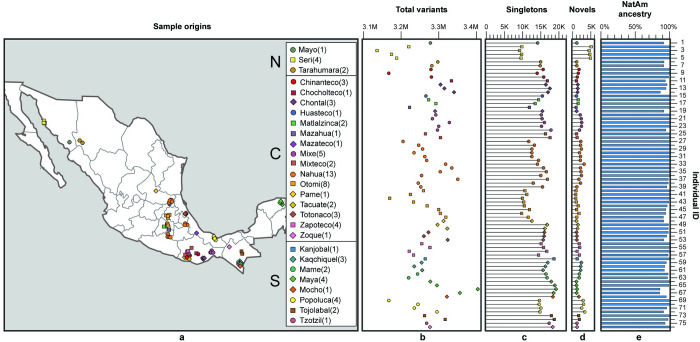
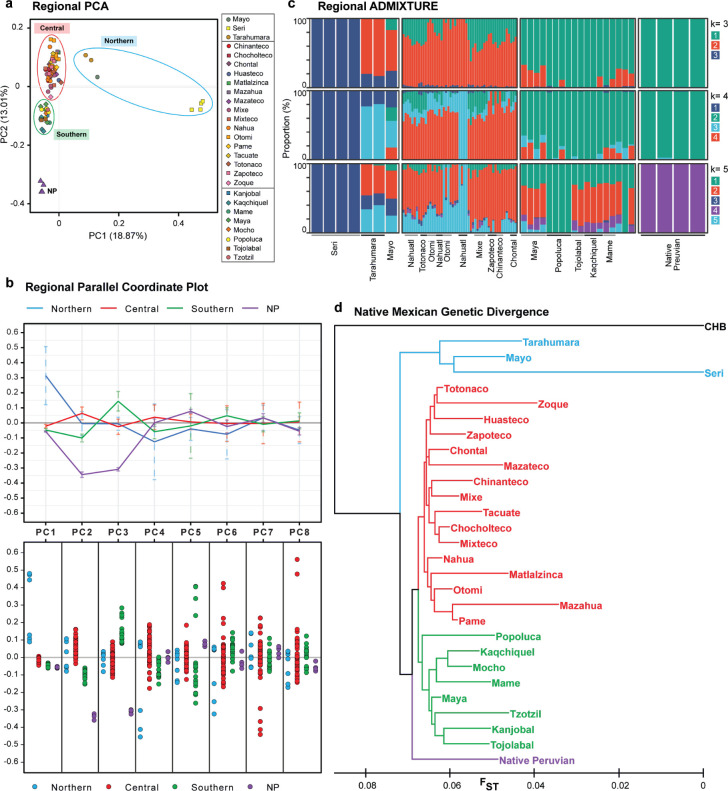
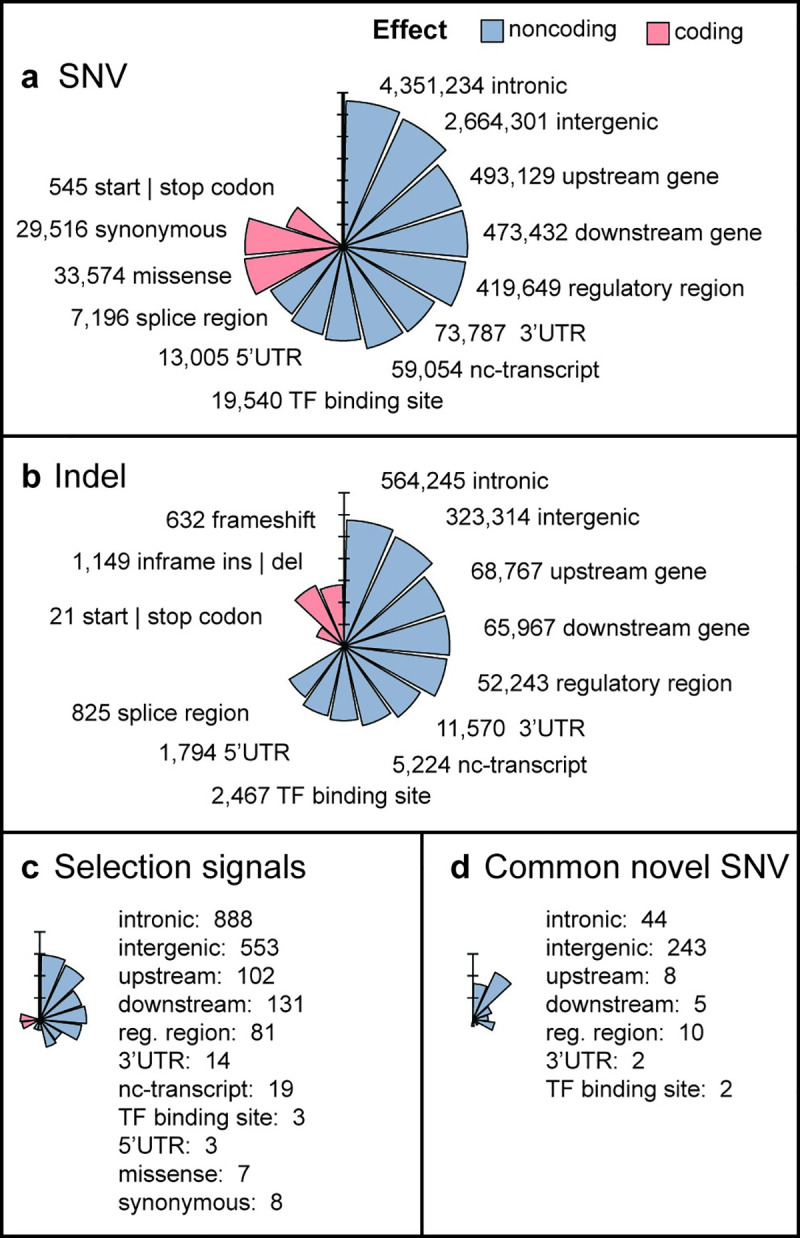
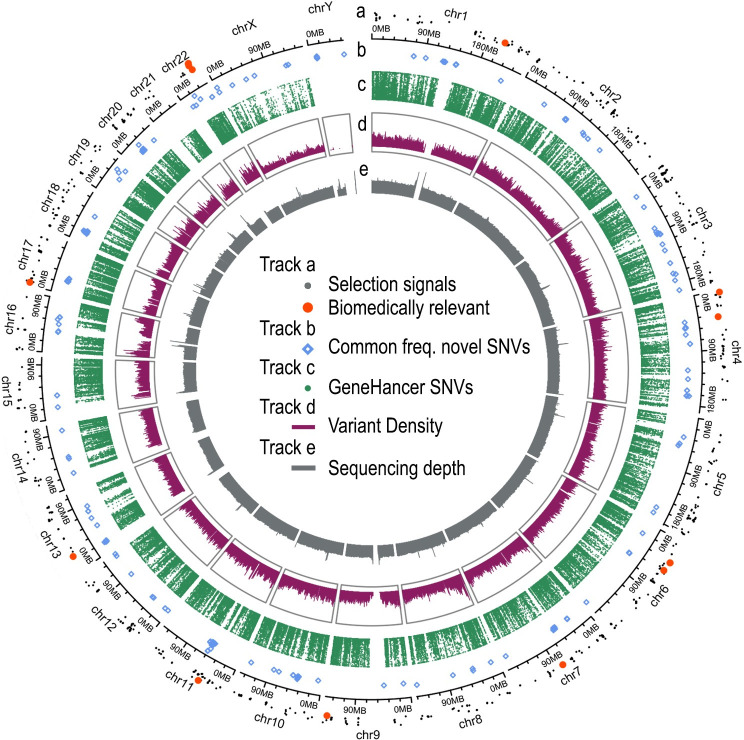
There has been limited study of Native American whole genome diversity to date, which impairs effective implementation of personalized medicine and a detailed description of its demographic history. Here we report high coverage whole genome sequencing of 76 unrelated individuals, from 27 indigenous groups across Mexico, with more than 97% average Native American ancestry. On average, each individual has 3.26 million Single Nucleotide Variants and short indels, that together comprise a catalog of 9,737,152 variants, 44,118 of which are novel. We report 497 common Single Nucleotide Variants (with allele frequency > 5%) mapped to drug responses and 316,577 in enhancer or promoter elements; interestingly we found some of these enhancer variants in PPARG, a nuclear receptor involved in highly prevalent health problems in Mexican population, such as obesity, diabetes, and insulin resistance. By detecting signals of positive selection we report 24 enriched key pathways under selection, most of them related to immune mechanisms. No missense variants in ACE2, the receptor responsible for the entry of the SARS CoV-2 virus, were found in any individual. Population genomics and phylogenetic analyses demonstrated stratification in a Northern-Central-Southern axis, with major substructure in the Central region. The Seri, a northern group with the most genetic divergence in our study, showed a distinctive genomic context with the most novel variants, and the most population specific genotypes. Genome-wide analysis showed that the average haplotype blocks are longer in Native Mexicans than in other world populations. With this dataset we describe previously undetected population level variation in Native Mexicans, helping to reduce the gap in genomic data representation of such groups.
迄今为止,对美洲原住民全基因组多样性的研究有限,这不利于个性化医疗的有效实施及其人口历史的详细描述。在此,我们报告了来自墨西哥27个土著群体的76名无亲缘关系个体的高覆盖全基因组测序结果,这些个体的美洲原住民血统平均超过97%。平均而言,每个个体有326万个单核苷酸变异和短插入缺失,这些变异共同构成了一个包含9737152个变异的目录,其中44118个是新变异。我们报告了497个映射到药物反应的常见单核苷酸变异(等位基因频率>5%)以及316577个存在于增强子或启动子元件中的变异;有趣的是,我们在PPARG中发现了一些此类增强子变异,PPARG是一种核受体,与墨西哥人群中高度普遍的健康问题有关,如肥胖、糖尿病和胰岛素抵抗。通过检测正选择信号,我们报告了24条在选择中富集的关键途径,其中大多数与免疫机制有关。在任何个体中均未发现负责SARS-CoV-2病毒进入的受体ACE2中的错义变异。群体基因组学和系统发育分析表明,在北-中-南轴上存在分层,中部地区存在主要亚结构。在我们的研究中,遗传差异最大的北部群体Seri显示出独特的基因组背景,具有最多的新变异和最具群体特异性的基因型。全基因组分析表明,墨西哥原住民的平均单倍型块比世界其他人群更长。利用这个数据集,我们描述了墨西哥原住民以前未被发现的群体水平变异,有助于缩小此类群体在基因组数据代表性方面的差距。