Wellcome Sanger Institute, Hinxton CB10 1SA, UK.
The Francis Crick Institute, London NW1 1AT, UK.
Science. 2020 Mar 20;367(6484). doi: 10.1126/science.aay5012.
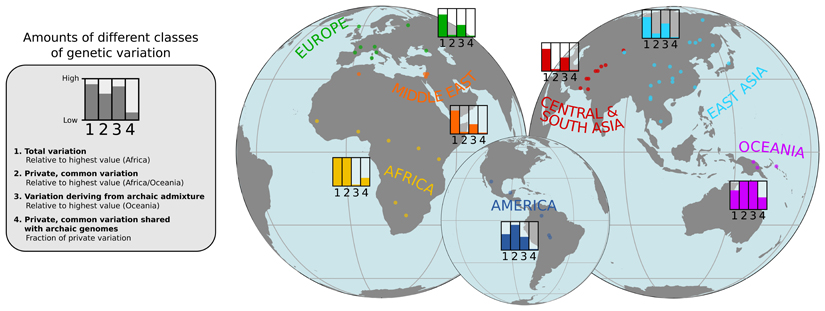
Genome sequences from diverse human groups are needed to understand the structure of genetic variation in our species and the history of, and relationships between, different populations. We present 929 high-coverage genome sequences from 54 diverse human populations, 26 of which are physically phased using linked-read sequencing. Analyses of these genomes reveal an excess of previously undocumented common genetic variation private to southern Africa, central Africa, Oceania, and the Americas, but an absence of such variants fixed between major geographical regions. We also find deep and gradual population separations within Africa, contrasting population size histories between hunter-gatherer and agriculturalist groups in the past 10,000 years, and a contrast between single Neanderthal but multiple Denisovan source populations contributing to present-day human populations.
需要来自不同人类群体的基因组序列来了解我们物种遗传变异的结构,以及不同人群之间的历史和关系。我们提供了 54 个不同人类群体的 929 个高覆盖率基因组序列,其中 26 个使用连接读取测序进行物理相位。对这些基因组的分析揭示了先前未记录的、仅存在于南非、中非、大洋洲和美洲的常见遗传变异的过剩,但不存在主要地理区域之间固定的此类变体。我们还发现非洲内部存在深刻而渐进的种群分离,过去 10000 年来,狩猎采集者和农业群体的人口规模历史存在差异,以及尼安德特人和丹尼索瓦人单一来源群体对现代人类群体的影响形成对比。