Wang Shining, Chen Grace, Merlo Pich Emilio, Affinito John, Cwik Michael, Faessel Hélène
Quantitative Clinical Pharmacology, Takeda Pharmaceuticals International Co, Cambridge, Massachusetts, USA.
Clinical Science, Takeda Pharmaceuticals International AG, Zurich, Switzerland.
Br J Clin Pharmacol. 2021 Nov;87(11):4354-4365. doi: 10.1111/bcp.14854. Epub 2021 May 5.
Soticlestat is a first-in-class selective inhibitor of cholesterol 24-hydroxylase, the enzyme that converts brain cholesterol to 24S-hydroxycholesterol (24HC), a positive allosteric modulator of N-methyl-D-aspartate receptors. Soticlestat is under development as treatment for rare developmental and epileptic encephalopathies.
In this first-in-human study, 48 healthy men and women received single ascending doses of soticlestat oral solution or placebo. Subsequently, nine healthy subjects received soticlestat tablets under fed and fasting conditions to assess the relative oral bioavailability and effects of food. Serial blood and urine samples were collected for pharmacokinetic and pharmacodynamic assessments.
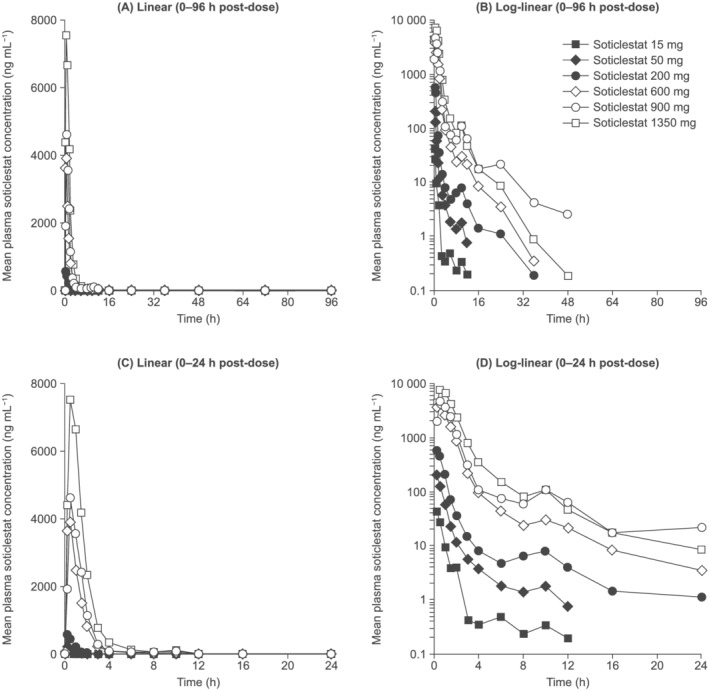
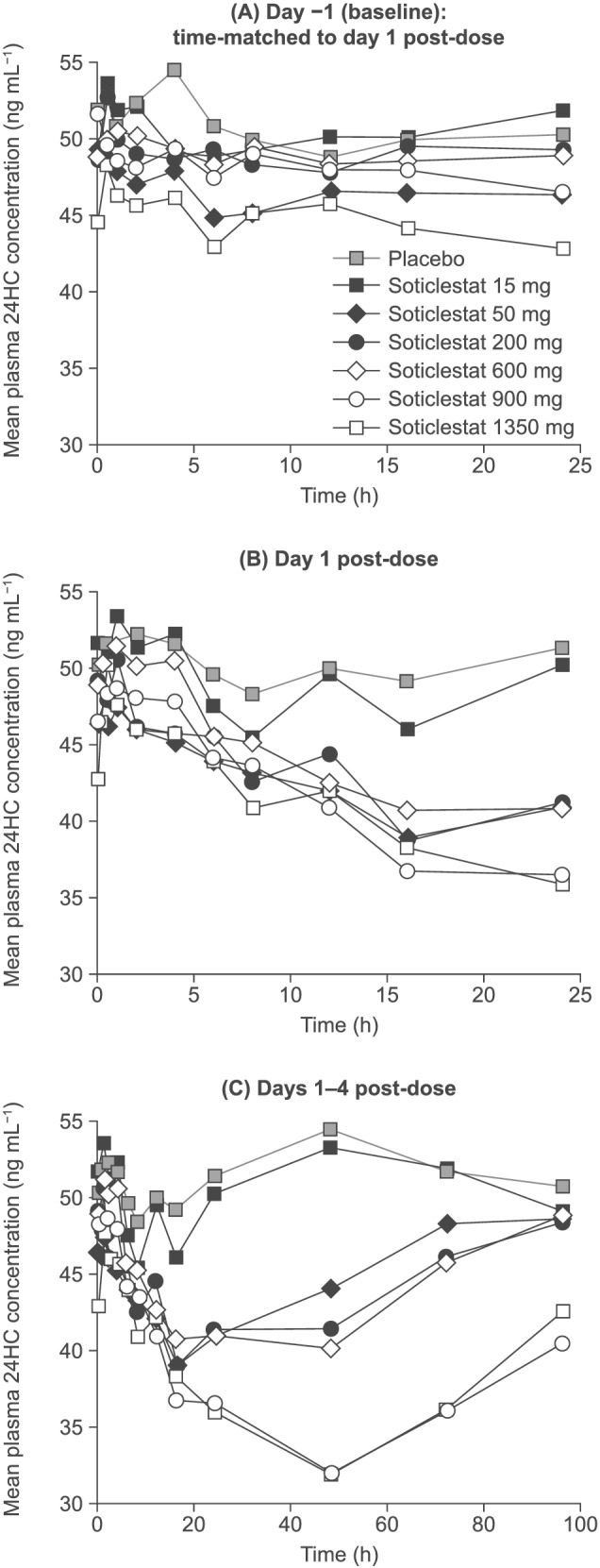
Soticlestat appeared to be well tolerated up to a single dose of 1350 mg. Adverse events (AEs) were mild in intensity, and dose-dependent increase in AE prevalence was not apparent. Soticlestat administered via oral solution was rapidly absorbed (median time to maximum plasma concentration [C ] 0.250-0.520 h). Mean C and area under plasma concentration-time curve from zero to infinity increased by 183- and 581-fold, respectively, over a 90-fold dose increase. Mean terminal elimination half-life was 0.820-7.16 hours across doses. Renal excretion was negligible. Administration of soticlestat tablets, and with food, lowered C but did not affect overall exposure. Plasma 24HC concentrations generally decreased with increasing dose.
Soticlestat appeared to be well tolerated after a single oral administration of up to 1350 mg and dose-dependently reduced plasma 24HC concentrations. Systemic exposure increased in a greater than dose-proportional manner over the dose range evaluated but was not affected by formulation or administration with food.
索替司他是一种一流的胆固醇24-羟化酶选择性抑制剂,该酶可将脑胆固醇转化为24S-羟胆固醇(24HC),而24HC是N-甲基-D-天冬氨酸受体的正变构调节剂。索替司他正处于开发阶段,用于治疗罕见的发育性和癫痫性脑病。
在这项首次人体研究中,48名健康男性和女性接受了索替司他口服溶液或安慰剂的单次递增剂量。随后,9名健康受试者在进食和空腹条件下服用索替司他片剂,以评估相对口服生物利用度和食物的影响。收集系列血液和尿液样本进行药代动力学和药效学评估。
索替司他单次剂量高达1350mg时耐受性良好。不良事件强度较轻,不良事件发生率未见剂量依赖性增加。通过口服溶液给药的索替司他吸收迅速(达到最大血浆浓度[C]的中位时间为0.250 - 0.520小时)。在剂量增加90倍的情况下,平均C和从零到无穷大的血浆浓度-时间曲线下面积分别增加了183倍和581倍。各剂量下平均终末消除半衰期为0.820 - 7.16小时。肾排泄可忽略不计。服用索替司他片剂以及与食物同服会降低C,但不影响总体暴露。血浆24HC浓度通常随剂量增加而降低。
单次口服高达1350mg的索替司他后耐受性良好,且剂量依赖性地降低血浆24HC浓度。在所评估的剂量范围内,全身暴露以大于剂量比例的方式增加,但不受制剂或与食物同服的影响。