Department of Genetics, Stanford University, Stanford, CA, USA.
Department of Biomedical Data Science, Stanford University, Stanford, CA, USA.
Nat Commun. 2021 Apr 12;12(1):2165. doi: 10.1038/s41467-021-22489-2.
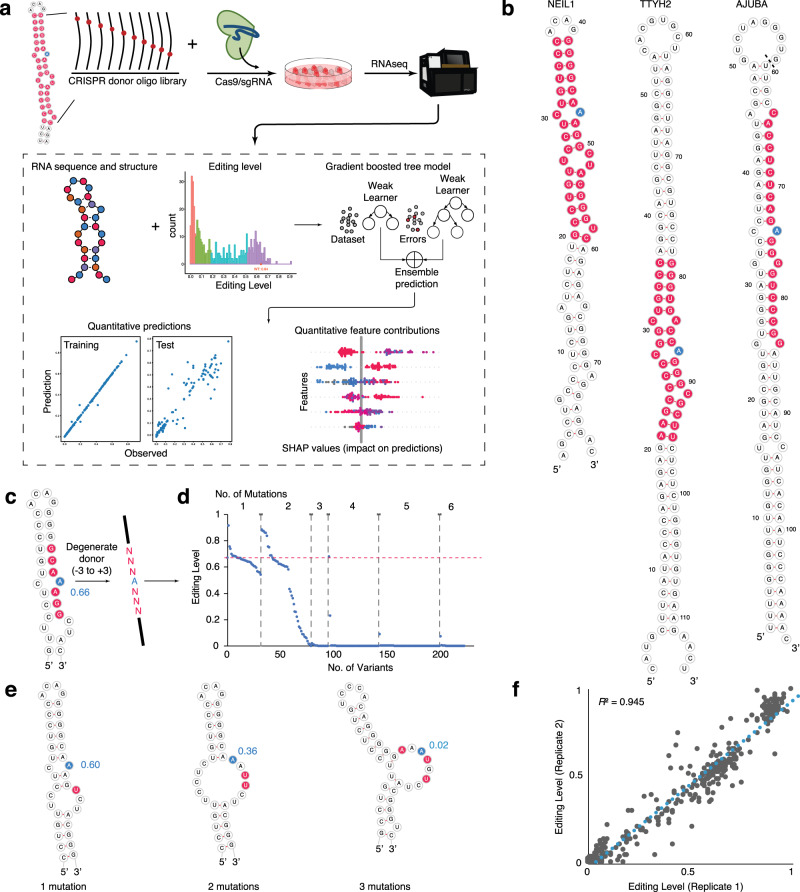
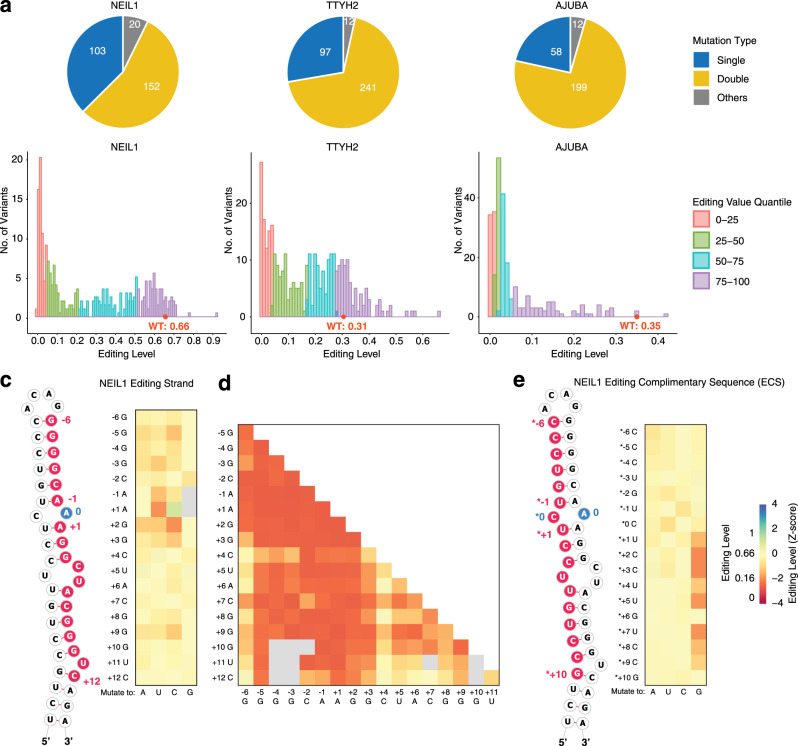
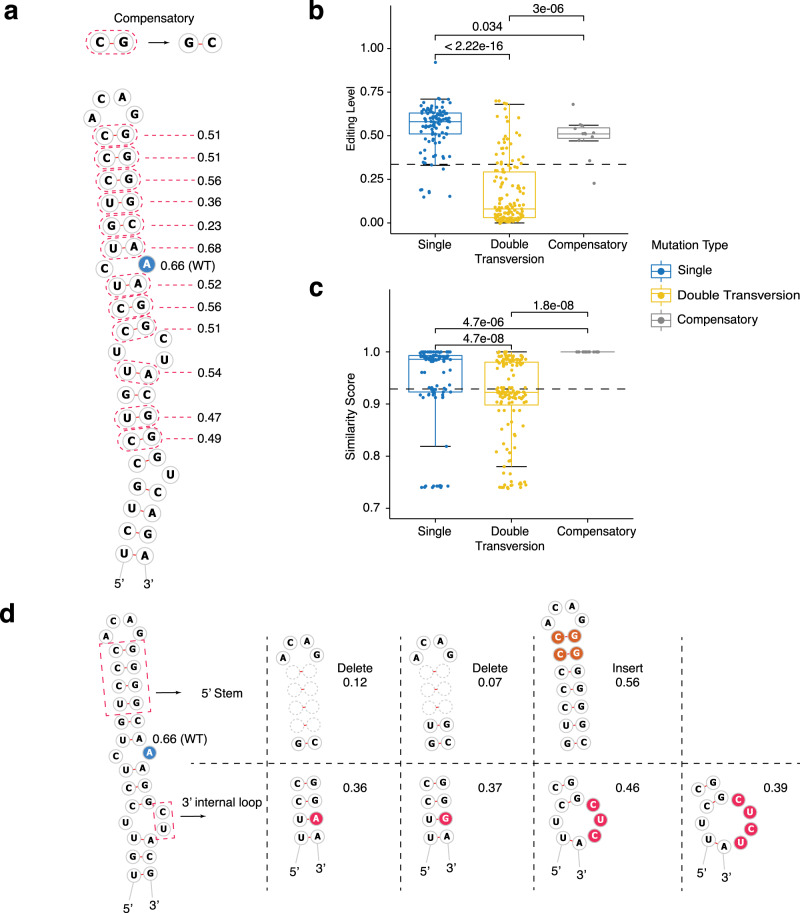
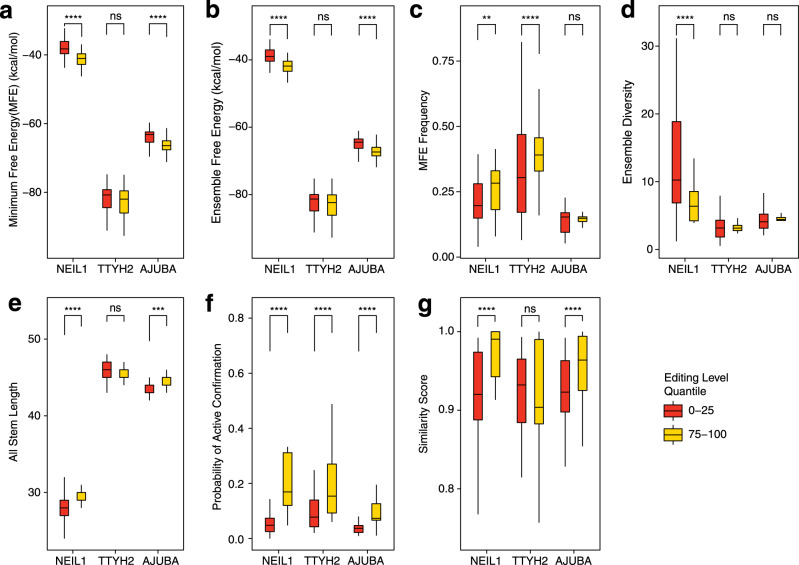
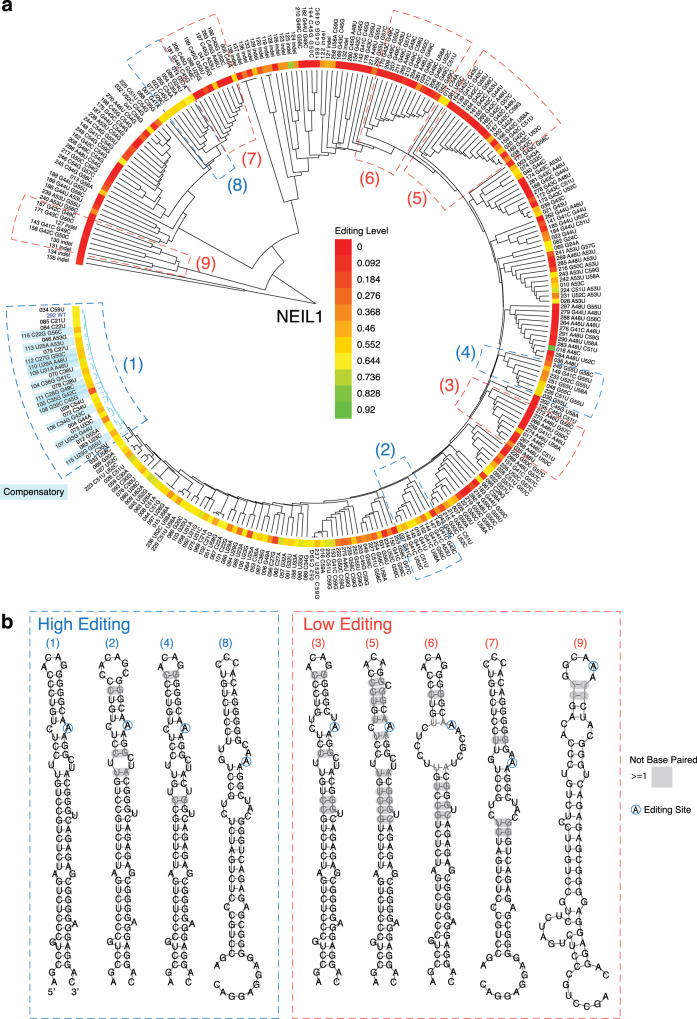
Adenosine-to-inosine (A-to-I) RNA editing catalyzed by ADAR enzymes occurs in double-stranded RNAs. Despite a compelling need towards predictive understanding of natural and engineered editing events, how the RNA sequence and structure determine the editing efficiency and specificity (i.e., cis-regulation) is poorly understood. We apply a CRISPR/Cas9-mediated saturation mutagenesis approach to generate libraries of mutations near three natural editing substrates at their endogenous genomic loci. We use machine learning to integrate diverse RNA sequence and structure features to model editing levels measured by deep sequencing. We confirm known features and identify new features important for RNA editing. Training and testing XGBoost algorithm within the same substrate yield models that explain 68 to 86 percent of substrate-specific variation in editing levels. However, the models do not generalize across substrates, suggesting complex and context-dependent regulation patterns. Our integrative approach can be applied to larger scale experiments towards deciphering the RNA editing code.
腺嘌呤到肌苷(A-to-I)RNA 编辑由 ADAR 酶催化发生在双链 RNA 中。尽管人们迫切需要对自然和工程编辑事件进行预测性理解,但 RNA 序列和结构如何决定编辑效率和特异性(即顺式调控)仍知之甚少。我们应用 CRISPR/Cas9 介导的饱和诱变方法,在其内源基因组位点附近的三个天然编辑底物上生成突变文库。我们使用机器学习整合不同的 RNA 序列和结构特征,通过深度测序来模拟编辑水平。我们确认了已知特征,并确定了对 RNA 编辑很重要的新特征。在同一底物内使用 XGBoost 算法进行训练和测试,得到的模型可以解释编辑水平的 68%到 86%的底物特异性变化。然而,这些模型不能跨底物推广,这表明存在复杂且依赖于上下文的调控模式。我们的综合方法可以应用于更大规模的实验,以破译 RNA 编辑密码。