EMBL-Rome, Epigenetics and Neurobiology Unit, Monterotondo (RM), Italy.
Blizard Institute, Centre for Genomics and Child Health, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, UK.
Commun Biol. 2021 Apr 12;4(1):478. doi: 10.1038/s42003-021-01944-2.
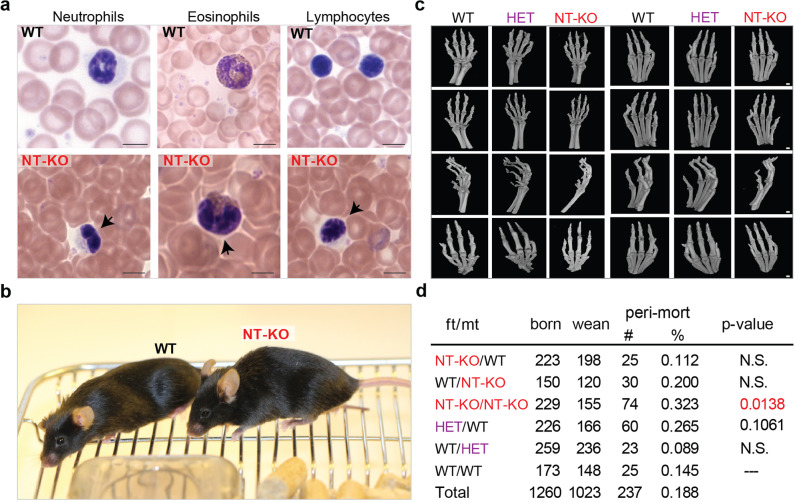
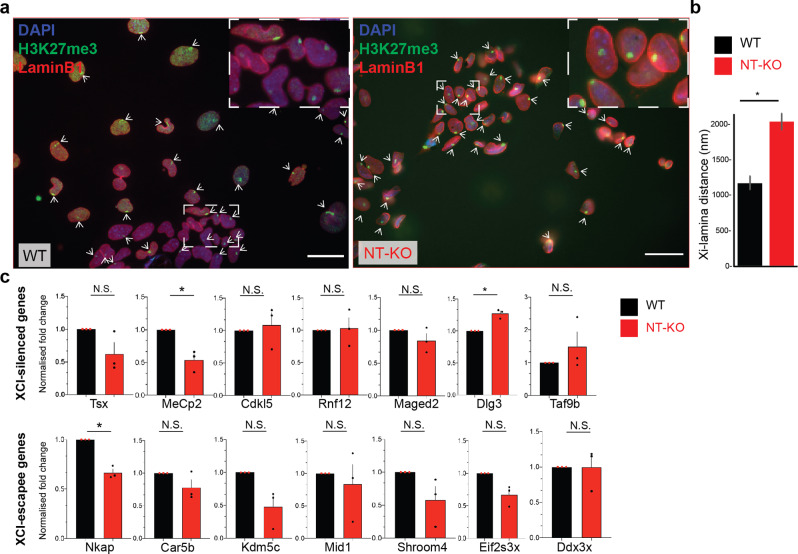
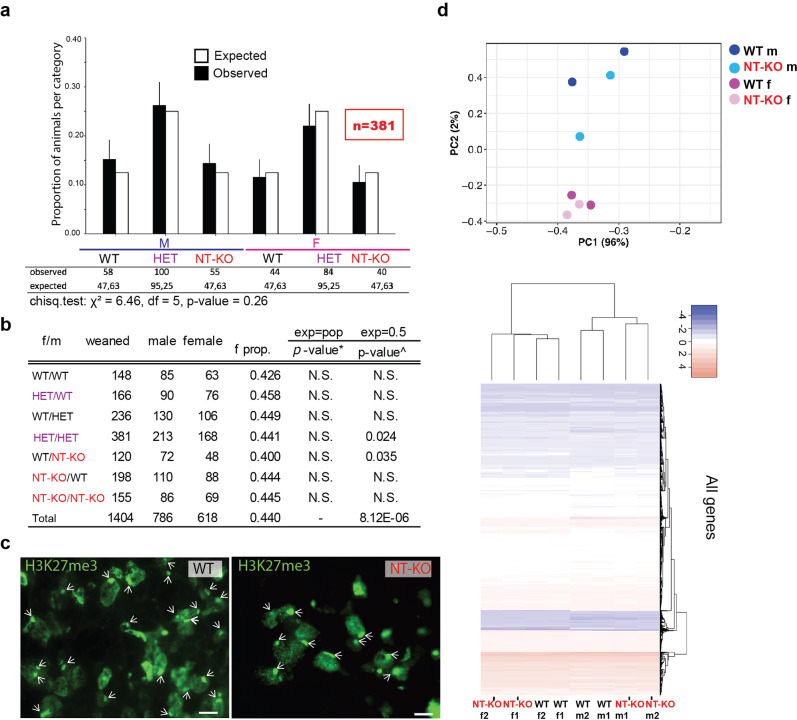
Mutations in the gene encoding Lamin B receptor (LBR), a nuclear-membrane protein with sterol reductase activity, have been linked to rare human disorders. Phenotypes range from a benign blood disorder, such as Pelger-Huet anomaly (PHA), affecting the morphology and chromatin organization of white blood cells, to embryonic lethality as for Greenberg dysplasia (GRBGD). Existing PHA mouse models do not fully recapitulate the human phenotypes, hindering efforts to understand the molecular etiology of this disorder. Here we show, using CRISPR/Cas-9 gene editing technology, that a 236bp N-terminal deletion in the mouse Lbr gene, generating a protein missing the N-terminal domains of LBR, presents a superior model of human PHA. Further, we address recent reports of a link between Lbr and defects in X chromosome inactivation (XCI) and show that our mouse mutant displays minor X chromosome inactivation defects that do not lead to any overt phenotypes in vivo. We suggest that our N-terminal deletion model provides a valuable pre-clinical tool to the research community and will aid in further understanding the etiology of PHA and the diverse functions of LBR.
编码核膜蛋白 Lamin B 受体(LBR)的基因突变与罕见的人类疾病有关。该基因具有固醇还原酶活性,其表型范围从良性血液疾病,如影响白细胞形态和染色质组织的Pelger-Huet 异常(PHA),到胚胎致死性的 Greenberg 发育不良(GRBGD)。现有的 PHA 小鼠模型不能完全再现人类表型,这阻碍了对该疾病分子病因的理解。在这里,我们使用 CRISPR/Cas-9 基因编辑技术,在小鼠 Lbr 基因中产生一个 236bp 的 N 端缺失,从而生成一个缺失 LBR N 端结构域的蛋白,这为人类 PHA 提供了一个更好的模型。此外,我们还针对最近关于 Lbr 与 X 染色体失活(XCI)缺陷之间的联系的报告进行了研究,并表明我们的小鼠突变体显示出轻微的 X 染色体失活缺陷,但在体内不会导致任何明显的表型。我们认为,我们的 N 端缺失模型为研究界提供了一个有价值的临床前工具,并将有助于进一步了解 PHA 的病因和 LBR 的多种功能。