Center for Cancer and Cell Biology, Van Andel Research Institute, Grand Rapids, MI, USA.
Department of Cellular and Molecular Medicine, University of Arizona Cancer Center, University of Arizona, Tucson, AZ, USA.
Oncogene. 2021 May;40(18):3260-3272. doi: 10.1038/s41388-021-01772-y. Epub 2021 Apr 12.
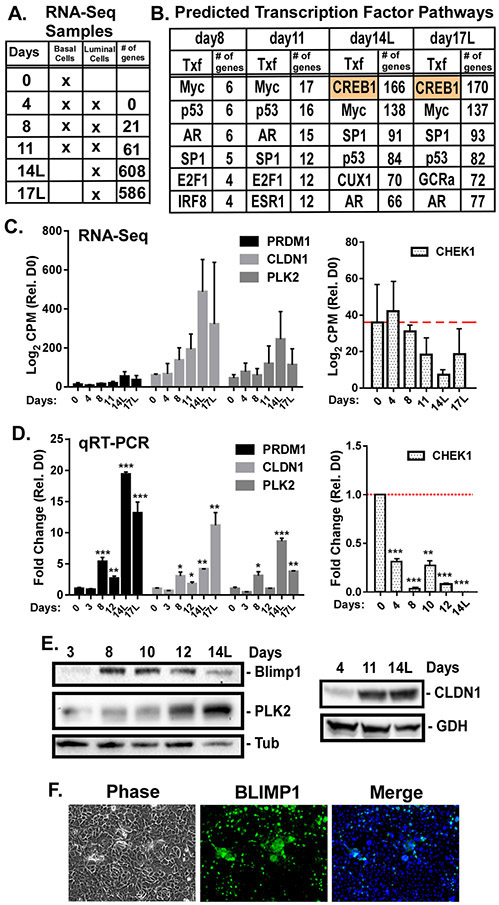
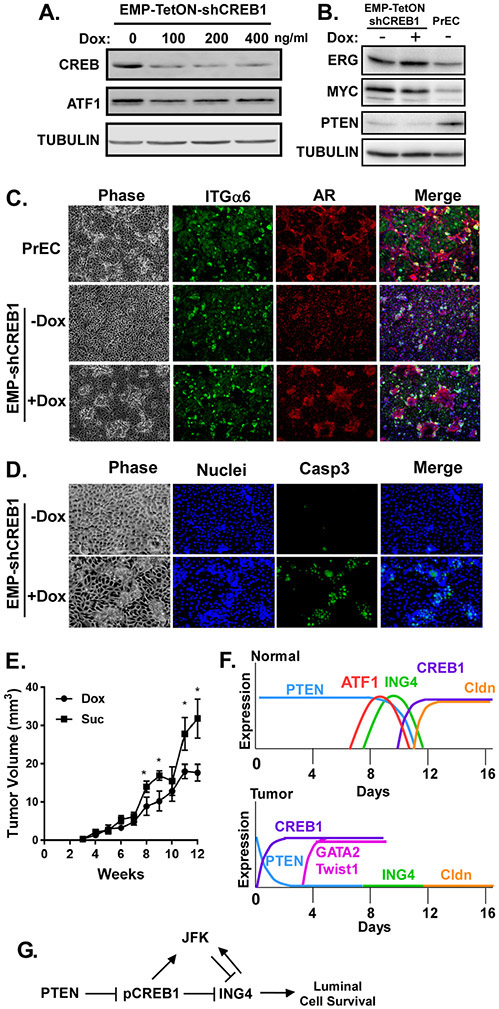
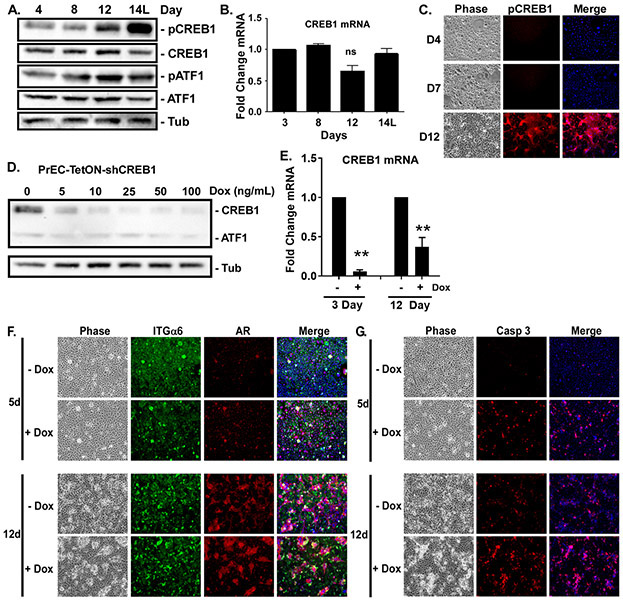
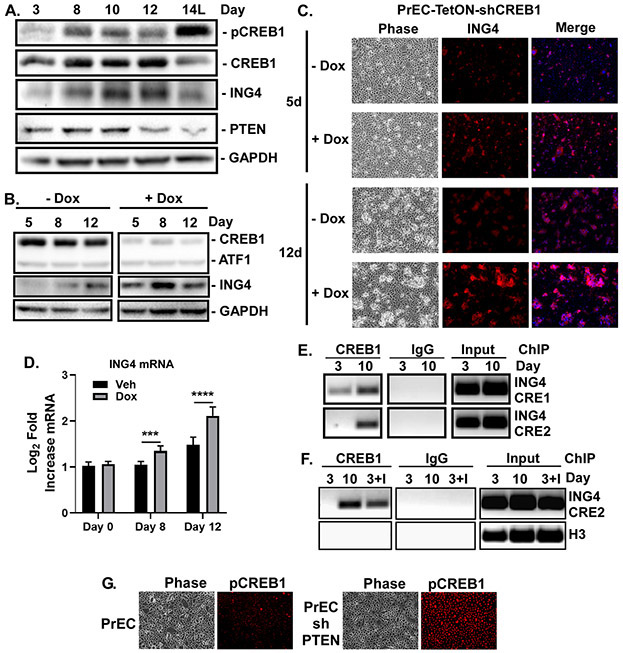
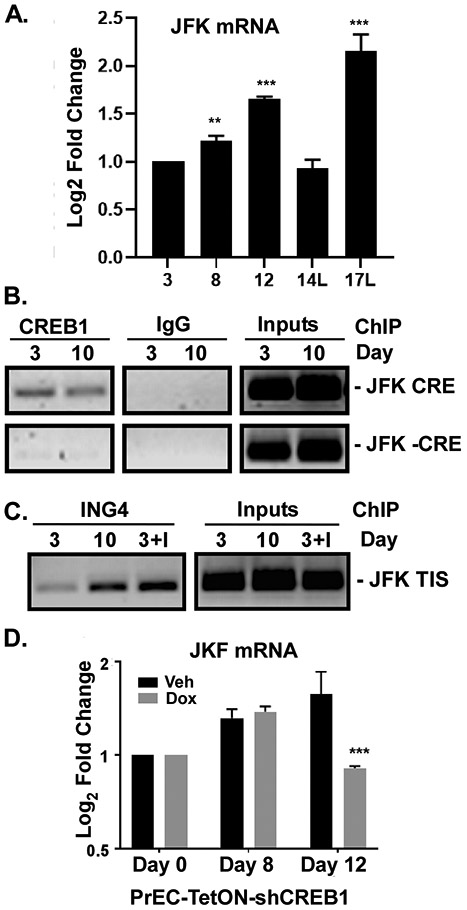
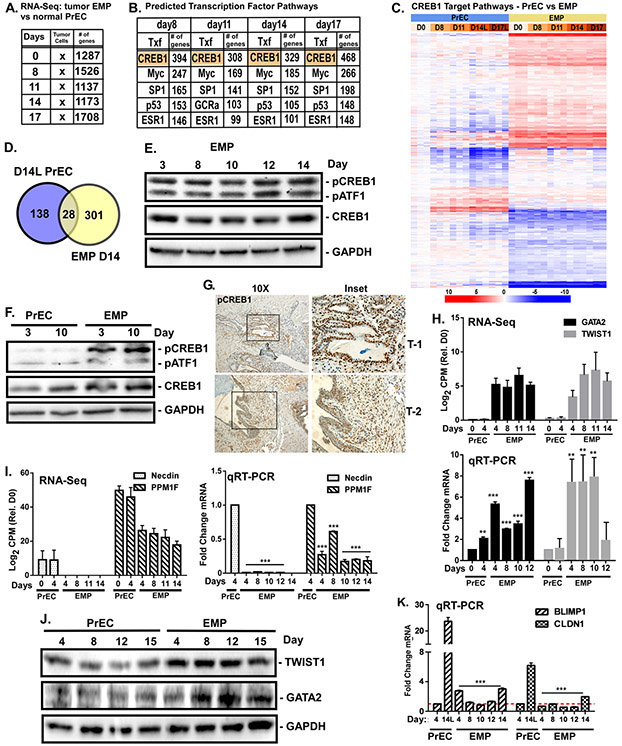
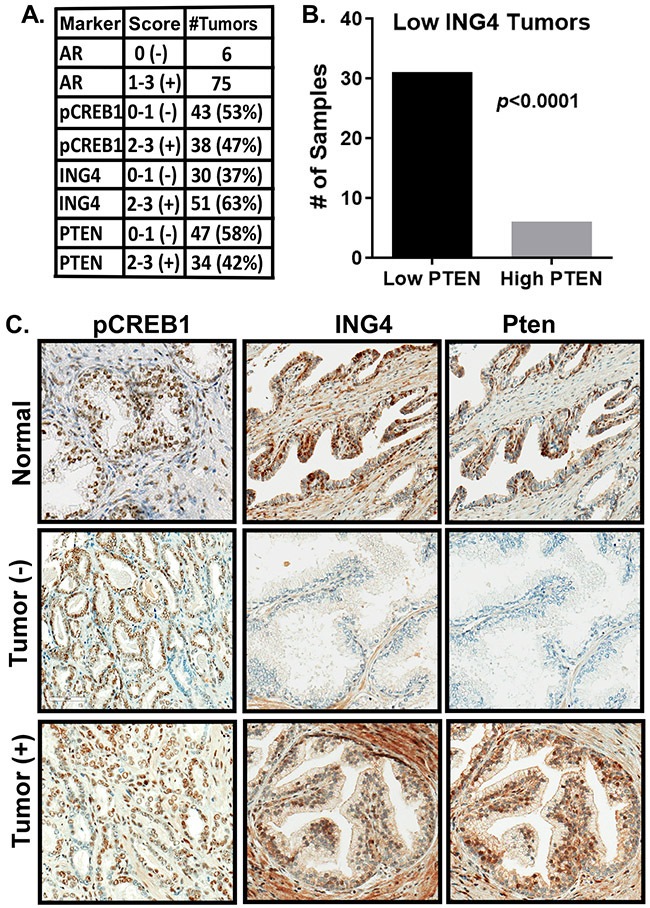
The molecular mechanisms of luminal cell differentiation are not understood well enough to determine how differentiation goes awry during oncogenesis. Using RNA-Seq analysis, we discovered that CREB1 plays a central role in maintaining new luminal cell survival and that oncogenesis dramatically changes the CREB1-induced transcriptome. CREB1 is active in luminal cells, but not basal cells. We identified ING4 and its E3 ligase, JFK, as CREB1 transcriptional targets in luminal cells. During luminal cell differentiation, transient induction of ING4 expression is followed by a peak in CREB1 activity, while JFK increases concomitantly with CREB1 activation. Transient expression of ING4 is required for luminal cell induction; however, failure to properly down-regulate ING4 leads to luminal cell death. Consequently, blocking CREB1 increased ING4 expression, suppressed JFK, and led to luminal cell death. Thus, CREB1 is responsible for the suppression of ING4 required for luminal cell survival and maintenance. Oncogenic transformation by suppressing PTEN resulted in constitutive activation of CREB1. However, the tumor cells could no longer fully differentiate into luminal cells, failed to express ING4, and displayed a unique CREB1 transcriptome. Blocking CREB1 in tumorigenic cells suppressed tumor growth in vivo, rescued ING4 expression, and restored luminal cell formation, but ultimately induced luminal cell death. IHC of primary prostate tumors demonstrated a strong correlation between loss of ING4 and loss of PTEN. This is the first study to define a molecular mechanism whereby oncogenic loss of PTEN, leading to aberrant CREB1 activation, suppresses ING4 expression causing disruption of luminal cell differentiation.
腔细胞分化的分子机制还没有被充分理解,因此无法确定肿瘤发生过程中分化是如何出错的。我们通过 RNA-Seq 分析发现,CREB1 在维持新的腔细胞存活方面发挥着核心作用,而肿瘤发生则显著改变了 CREB1 诱导的转录组。CREB1 在腔细胞中活跃,但不在基底细胞中活跃。我们鉴定出 ING4 及其 E3 连接酶 JFK 是腔细胞中 CREB1 的转录靶点。在腔细胞分化过程中,ING4 表达的短暂诱导后紧跟着 CREB1 活性的峰值,而 JFK 则随着 CREB1 的激活而同时增加。ING4 的短暂表达是诱导腔细胞所必需的;然而,ING4 没有被正确下调会导致腔细胞死亡。因此,阻断 CREB1 会增加 ING4 的表达,抑制 JFK,并导致腔细胞死亡。因此,CREB1 负责抑制 ING4,这是腔细胞存活和维持所必需的。通过抑制 PTEN 进行的致癌转化导致 CREB1 的持续激活。然而,肿瘤细胞再也不能完全分化为腔细胞,无法表达 ING4,并表现出独特的 CREB1 转录组。在肿瘤细胞中阻断 CREB1 抑制了体内肿瘤的生长,挽救了 ING4 的表达,并恢复了腔细胞的形成,但最终诱导了腔细胞的死亡。对原发性前列腺肿瘤的免疫组化分析表明,ING4 的缺失与 PTEN 的缺失之间存在很强的相关性。这是第一项定义分子机制的研究,该机制表明,致癌性的 PTEN 丧失导致异常的 CREB1 激活,抑制 ING4 的表达,从而破坏腔细胞分化。