CNRS UMR7275, Institut de Pharmacologie Moléculaire et Cellulaire (IPMC), Université Côte d'Azur, Valbonne, France.
INSERM U1068, Centre de Recherche en Cancérologie de Marseille, Equipe Oncologie Prédictive, Aix-Marseille Université UM105, Marseille, France.
Nat Commun. 2021 Apr 13;12(1):2198. doi: 10.1038/s41467-021-22522-4.
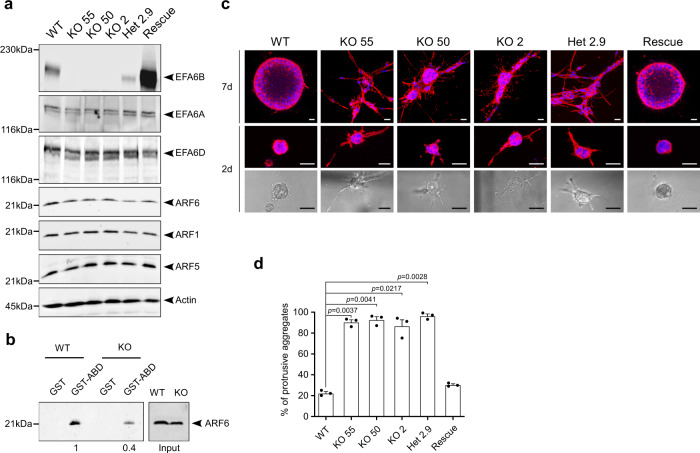
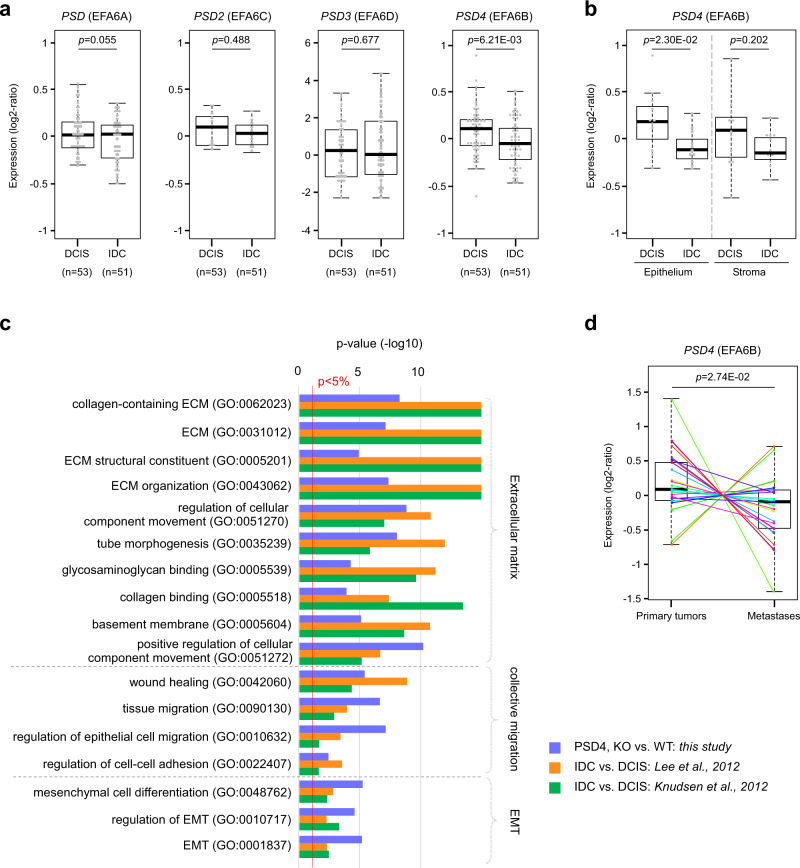
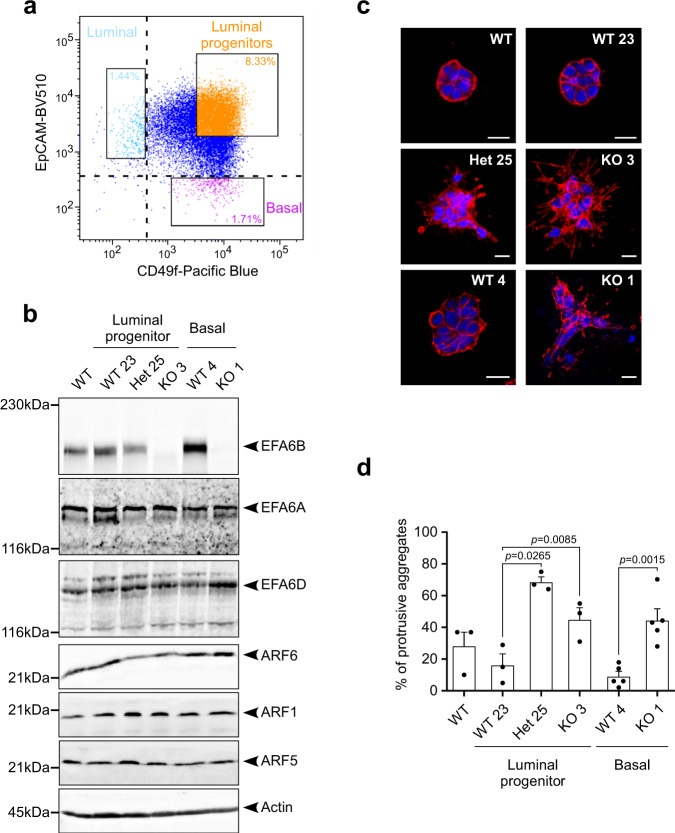
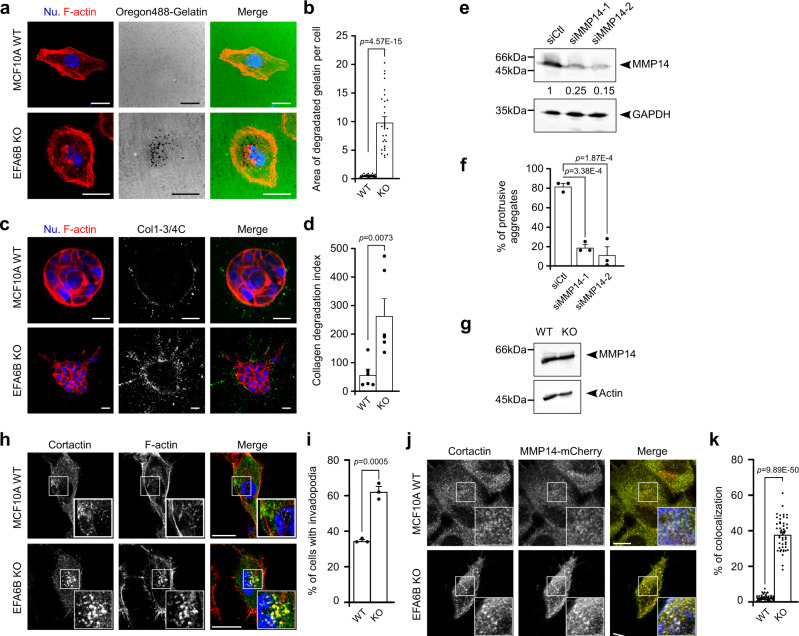
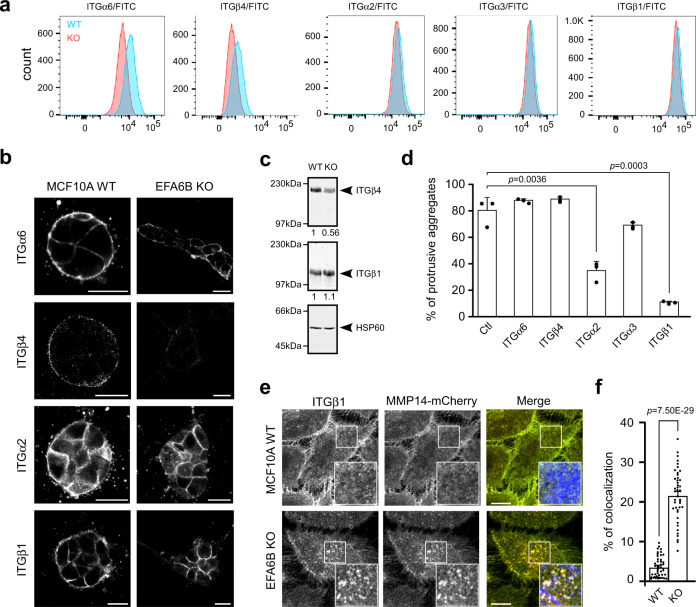
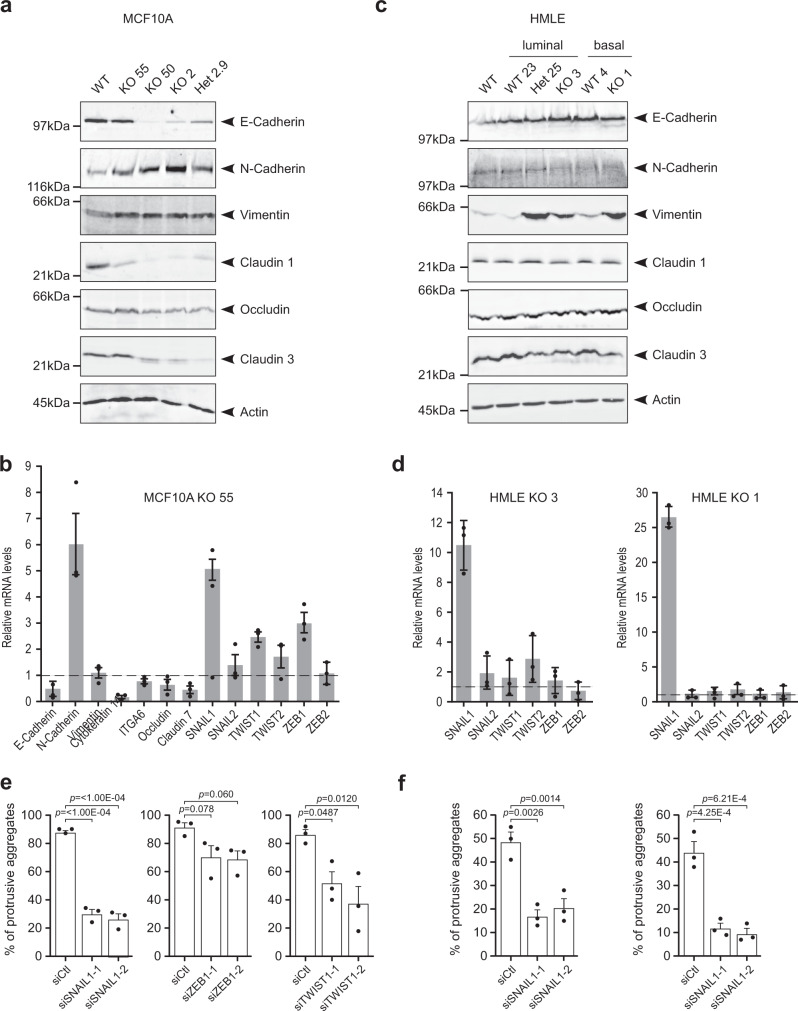
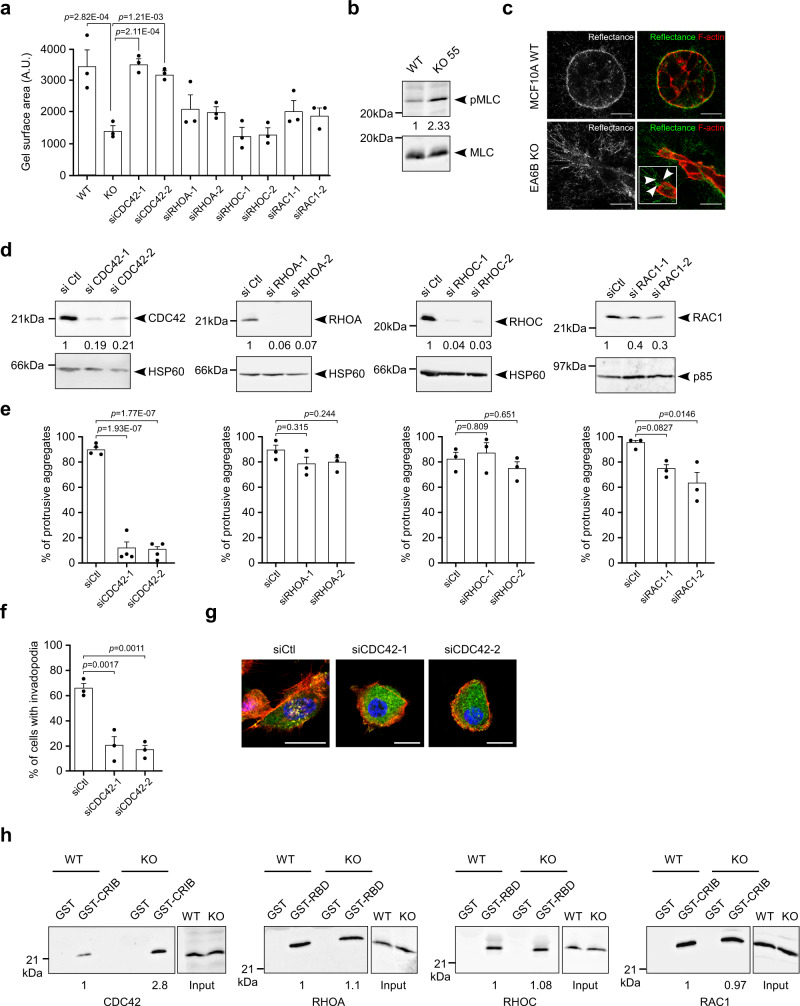
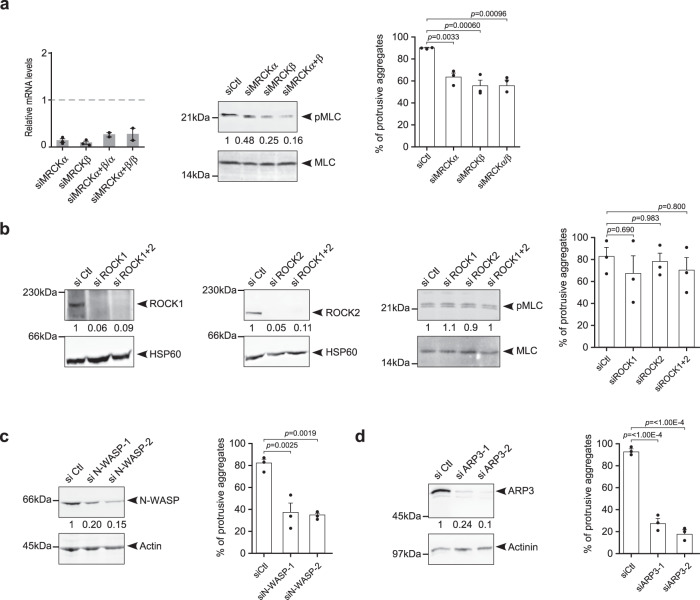
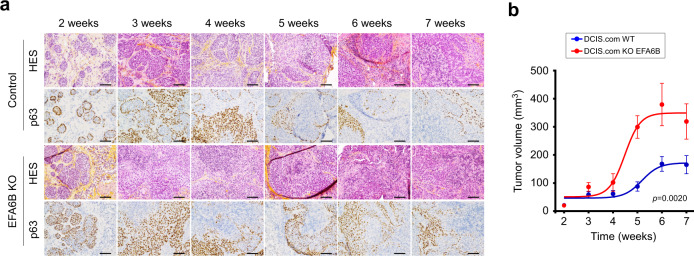
Cancer is initiated by somatic mutations in oncogenes or tumor suppressor genes. However, additional alterations provide selective advantages to the tumor cells to resist treatment and develop metastases. Their identification is of paramount importance. Reduced expression of EFA6B (Exchange Factor for ARF6, B) is associated with breast cancer of poor prognosis. Here, we report that loss of EFA6B triggers a transcriptional reprogramming of the cell-to-ECM interaction machinery and unleashes CDC42-dependent collective invasion in collagen. In xenograft experiments, MCF10 DCIS.com cells, a DCIS-to-IDC transition model, invades faster when knocked-out for EFA6B. In addition, invasive and metastatic tumors isolated from patients have lower expression of EFA6B and display gene ontology signatures identical to those of EFA6B knock-out cells. Thus, we reveal an EFA6B-regulated molecular mechanism that controls the invasive potential of mammary cells; this finding opens up avenues for the treatment of invasive breast cancer.
癌症是由癌基因或肿瘤抑制基因的体细胞突变引发的。然而,其他的改变为肿瘤细胞提供了选择性优势,使其能够抵抗治疗并发展转移。因此,识别这些改变至关重要。EFA6B(ARF6 的交换因子,B)表达降低与预后不良的乳腺癌有关。在这里,我们报告称,EFA6B 的缺失会触发细胞与细胞外基质相互作用机制的转录重编程,并释放 CDC42 依赖性的胶原集体侵袭。在异种移植实验中,MCF10 DCIS.com 细胞,一种 DCIS 到 IDC 转化模型,当 EFA6B 被敲除时,侵袭速度更快。此外,从患者中分离出的侵袭性和转移性肿瘤的 EFA6B 表达水平较低,并且表现出与 EFA6B 敲除细胞相同的基因本体论特征。因此,我们揭示了一个由 EFA6B 调节的分子机制,该机制控制着乳腺细胞的侵袭潜力;这一发现为治疗侵袭性乳腺癌开辟了新的途径。