Ding Yi, Li Min, Tayier Tuersong, Zhang MeiLin, Chen Long, Feng ShuMei
Department of Histology and Embryology, School of Basic Medical Sciences, Xinjiang Medical University, Urumqi, Xinjiang, China.
Department of Pharmacology, Pharmacy College, Xinjiang Medical University, Urumqi, China.
J Cancer. 2021 Mar 15;12(10):2921-2932. doi: 10.7150/jca.51851. eCollection 2021.
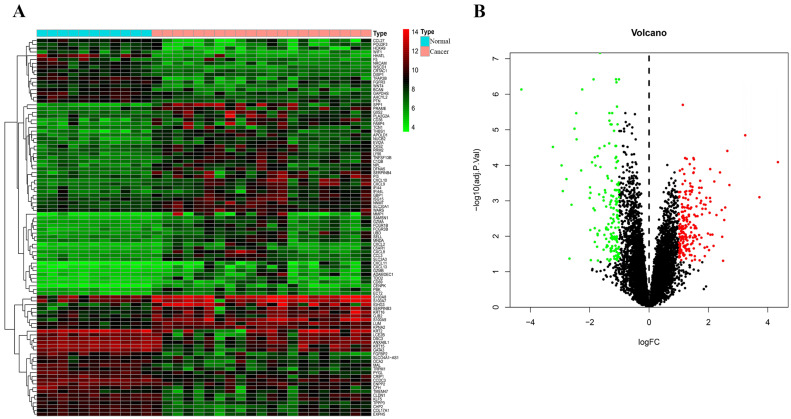
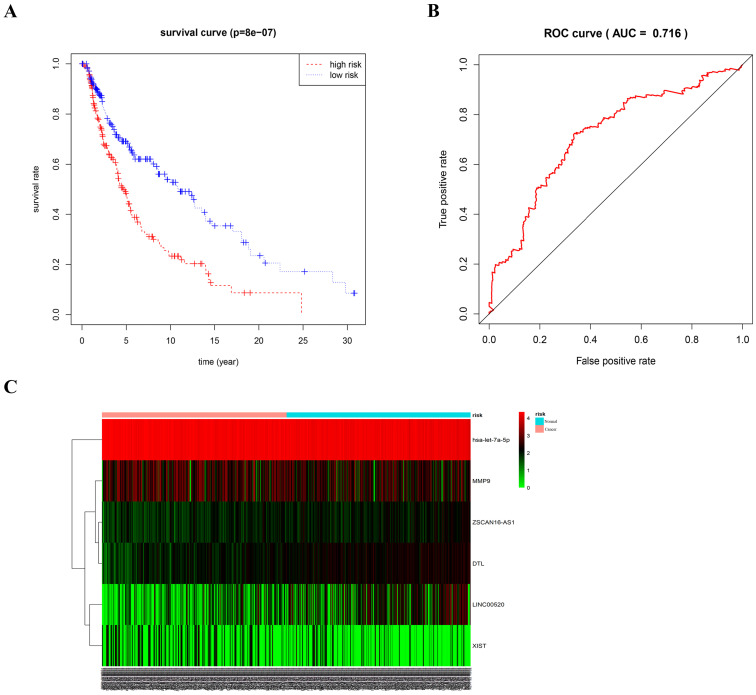
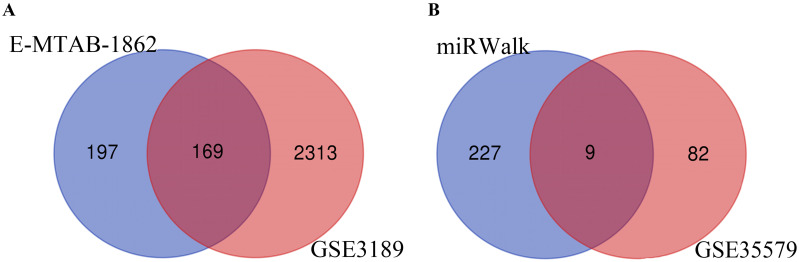
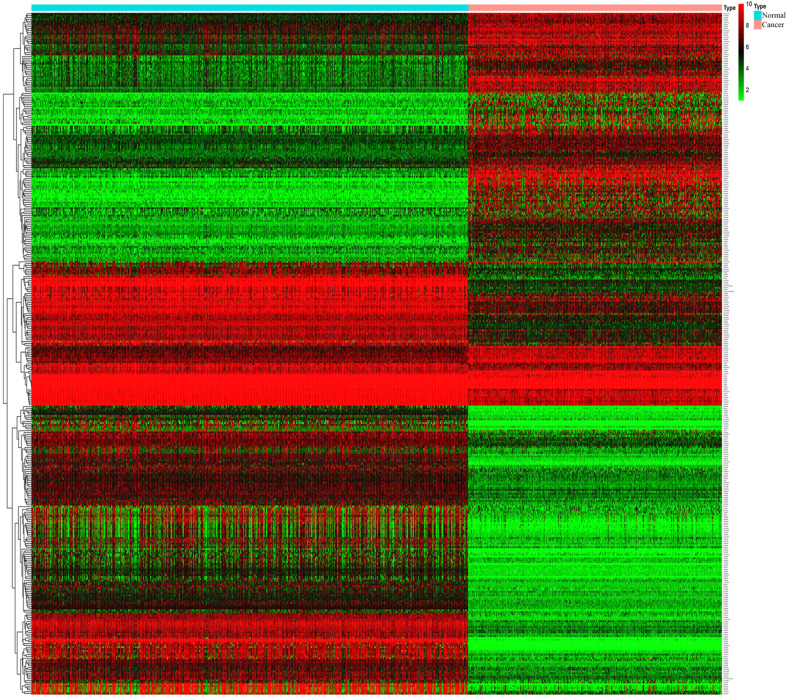
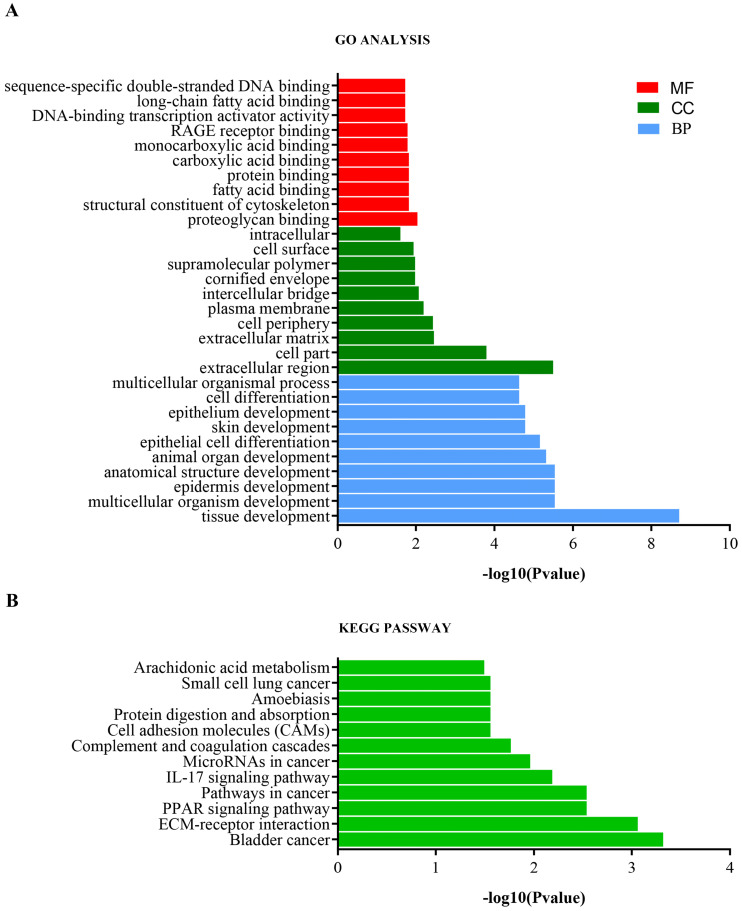
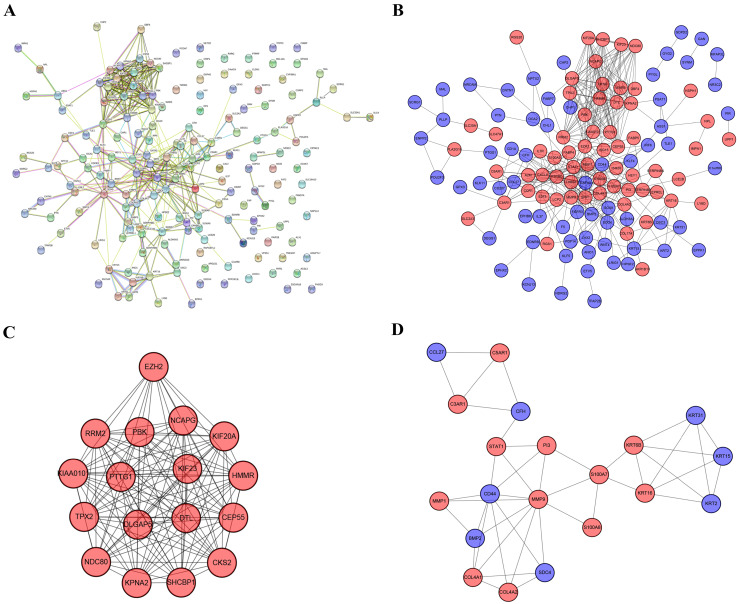
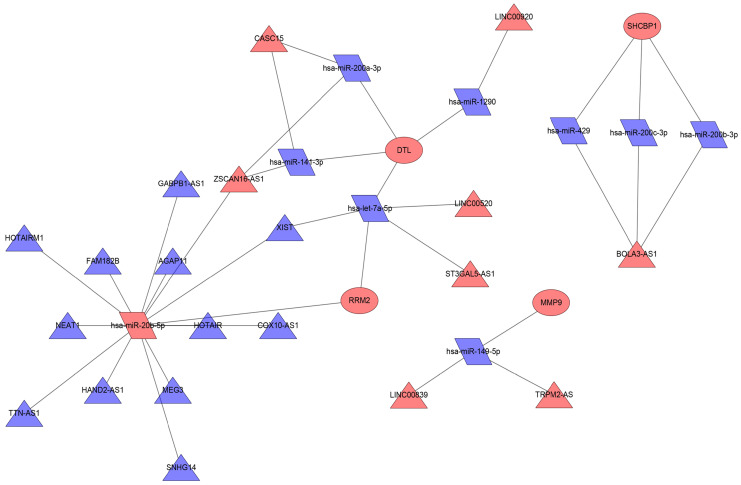
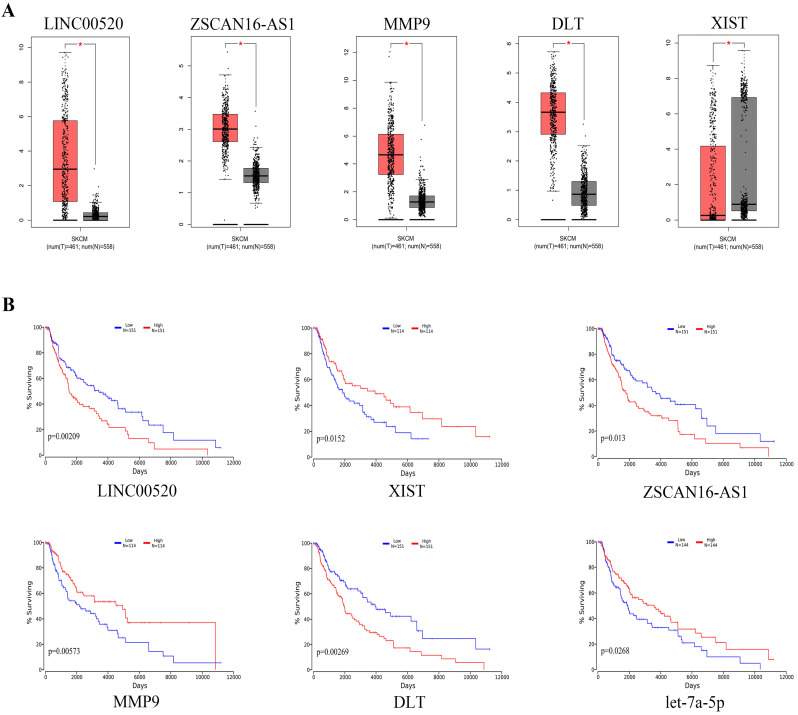
Melanoma is an extremely malignant tumor with early metastasis and high mortality. Little is known about the process of by which melanoma occurs, as its mechanism is very complex and only limited data are available on its long non-coding RNA (lncRNA)-associated competing endogenous RNAs (ceRNAs). The purpose of this study was to screen out potential prognostic molecules and identify a ceRNA network related to the occurrence of melanoma. We screened 169 differentially expressed mRNAs (DEmRNAs) from E-MTAB-1862 and GSE3189; gene ontology (GO) enrichment analysis showed that these genes were closely related to the development of skin. In the protein-protein interaction network, we screened out a total of 19 hub genes. Furthermore, we predicted the microRNAs (miRNAs) that regulate hub genes using the miRWalk database and then intersected these with GSE35579, resulting in nine DEmiRNAs. We also predicted the lncRNAs that regulate the miRNAs using the LncBasev.2 database. According to the ceRNA hypothesis, and based on the intersection of the DElncRNAs with merged GTEx and TCGA data, we obtained 20 DElncRNAs. A total of four DEmRNAs, nine DEmiRNAs, and 20 DElncRNAs were included in the ceRNA network. Based on Cox stepwise regression and survival analysis, we identified five biomarkers, ZSCAN16-AS1, LINC00520, XIST, DTL, and let-7a-5p, and obtained risk scores. The results showed that most of the differentially expressed genes were related to epithelial-mesenchymal transition (EMT) in melanoma. Finally, we obtained a LINC00520/let-7a-5p/DTL molecular regulatory network. These results suggest that ceRNA networks have an important role in evaluating the prognosis of patients with melanoma and provide a new experimental basis for exploring the EMT process in the development of melanoma.
黑色素瘤是一种极具恶性的肿瘤,具有早期转移和高死亡率。由于其发生机制非常复杂,且关于其长链非编码RNA(lncRNA)相关的竞争性内源RNA(ceRNA)的数据有限,人们对黑色素瘤的发生过程知之甚少。本研究的目的是筛选出潜在的预后分子,并鉴定与黑色素瘤发生相关的ceRNA网络。我们从E-MTAB-1862和GSE3189中筛选出169个差异表达的mRNA(DEmRNA);基因本体(GO)富集分析表明,这些基因与皮肤发育密切相关。在蛋白质-蛋白质相互作用网络中,我们共筛选出19个枢纽基因。此外,我们使用miRWalk数据库预测调控枢纽基因的微小RNA(miRNA),然后将这些miRNA与GSE35579进行交叉分析,得到9个差异表达的miRNA(DEmiRNA)。我们还使用LncBasev.2数据库预测调控这些miRNA的lncRNA。根据ceRNA假说,并基于差异表达lncRNA(DElncRNA)与合并的GTEx和TCGA数据的交叉分析,我们获得了20个DElncRNA。ceRNA网络共包含4个DEmRNA、9个DEmiRNA和20个DElncRNA。基于Cox逐步回归和生存分析,我们鉴定出5个生物标志物,即ZSCAN16-AS1、LINC00520、XIST、DTL和let-7a-5p,并获得了风险评分。结果表明,大多数差异表达基因与黑色素瘤的上皮-间质转化(EMT)有关。最后,我们获得了一个LINC00520/let-7a-5p/DTL分子调控网络。这些结果表明,ceRNA网络在评估黑色素瘤患者的预后中具有重要作用,并为探索黑色素瘤发生发展过程中的EMT过程提供了新的实验依据。