Tian Feng, Zhang Ying
Department of Cardiology, The First Medical Center of PLA General Hospital, Beijing, China.
Front Cell Dev Biol. 2021 Apr 1;9:636553. doi: 10.3389/fcell.2021.636553. eCollection 2021.
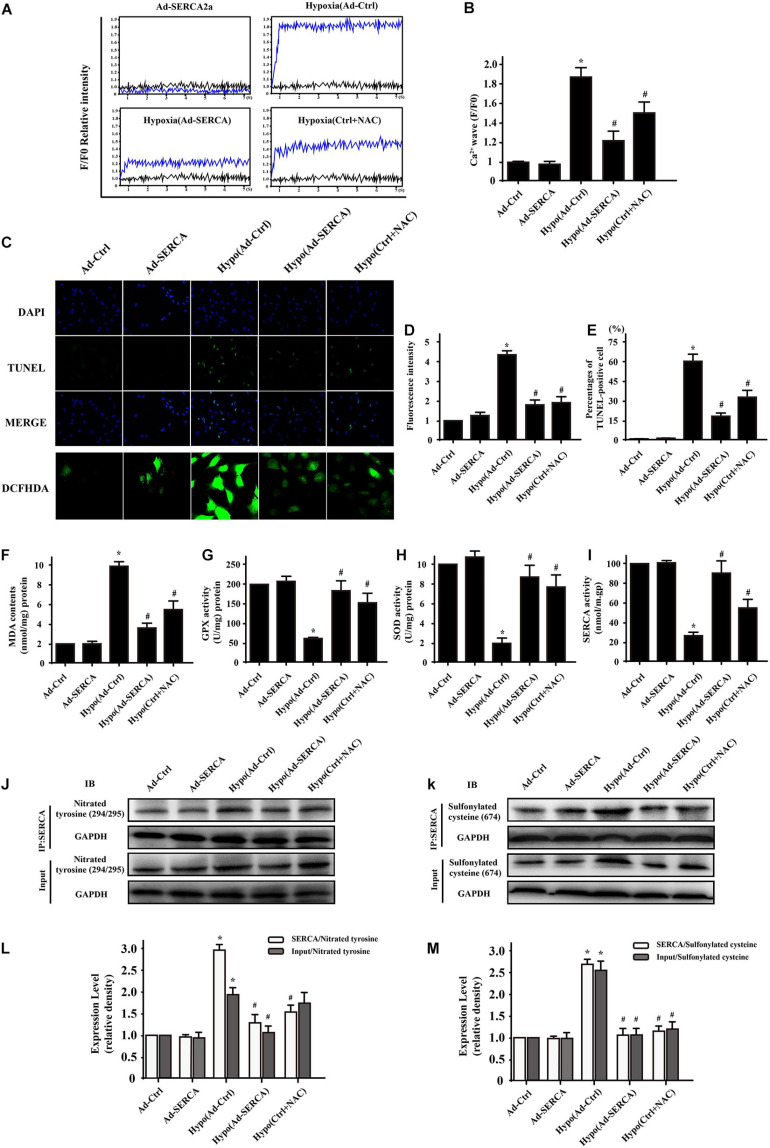
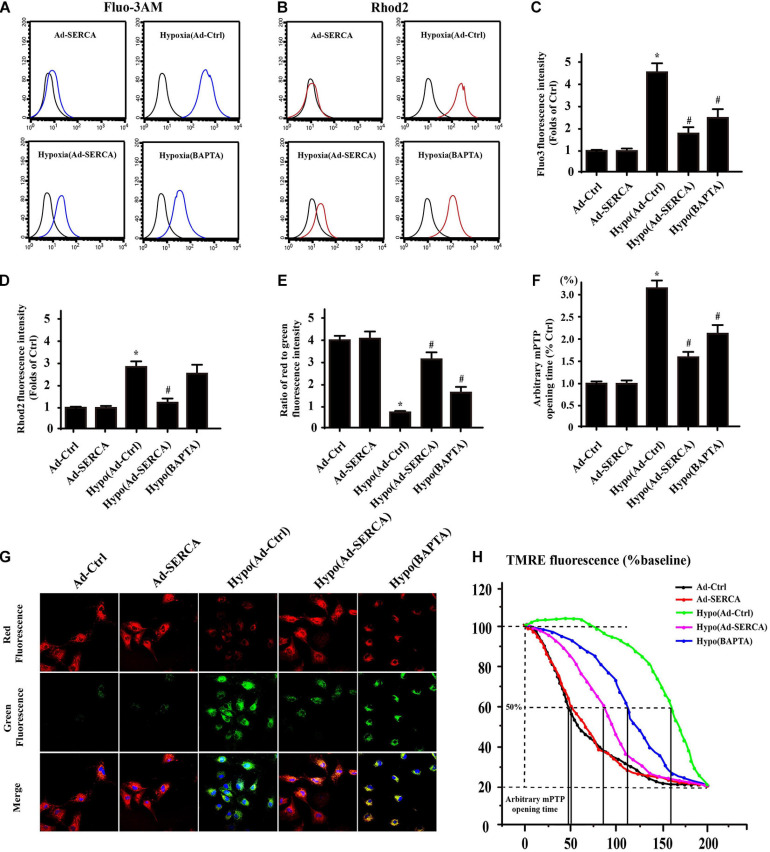
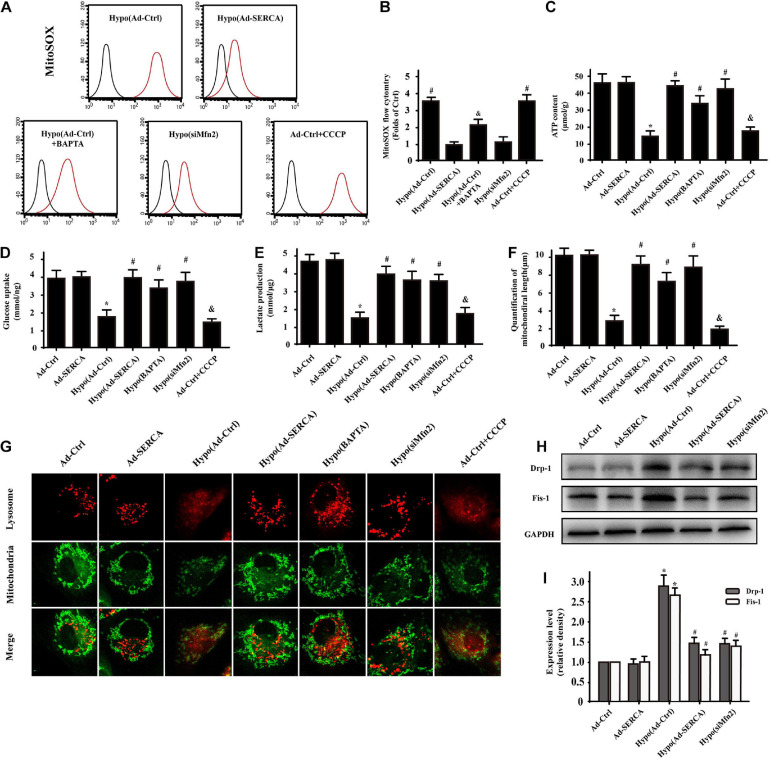
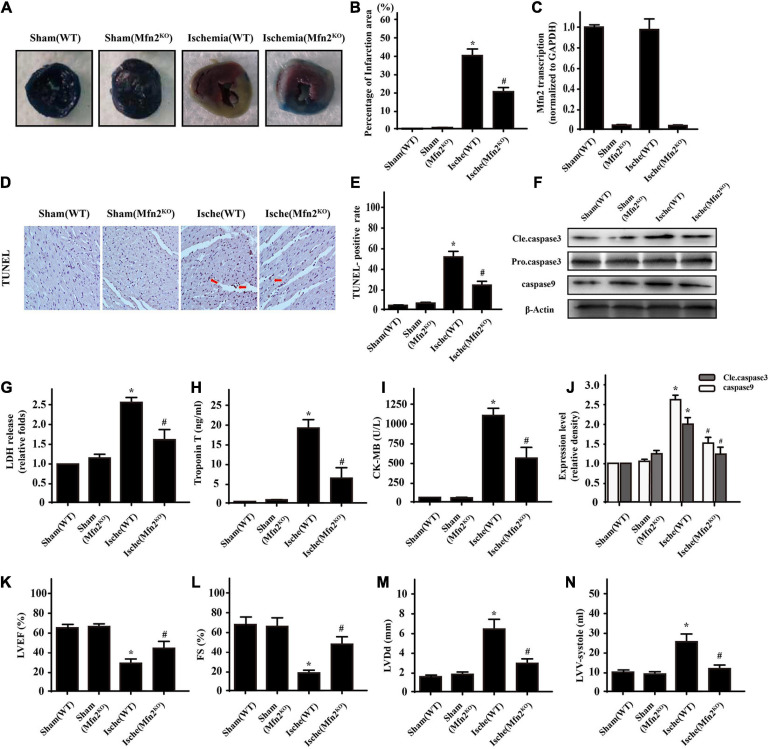
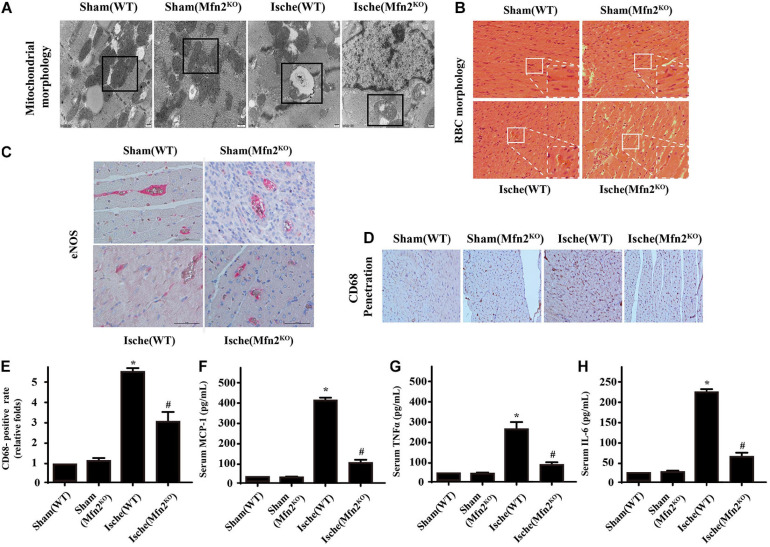
Our previous research has shown that type-2a Sarco/endoplasmic reticulum Ca-ATPase (SERCA2a) undergoes posttranscriptional oxidative modifications in cardiac microvascular endothelial cells (CMECs) in the context of excessive cardiac oxidative injury. However, whether SERCA2a inactivity induces cytosolic Ca imbalance in mitochondrial homeostasis is far from clear. Mitofusin2 (Mfn2) is well known as an important protein involved in endoplasmic reticulum (ER)/mitochondrial Ca tethering and the regulation of mitochondrial quality. Therefore, the aim of our study was to elucidate the specific mechanism of SERCA2a-mediated Ca overload in the mitochondria via Mfn2 tethering and the survival rate of the heart under conditions of cardiac microvascular ischemic injury. , CMECs extracted from mice were subjected to 6 h of hypoxic injury to mimic ischemic heart injury. C57-WT and Mfn2 mice were subjected to a 1 h ischemia procedure via ligation of the left anterior descending branch to establish an cardiac ischemic injury model. TTC staining, immunohistochemistry and echocardiography were used to assess the myocardial infarct size, microvascular damage, and heart function. , ischemic injury induced irreversible oxidative modification of SERCA2a, including sulfonylation at cysteine 674 and nitration at tyrosine 294/295, and inactivation of SERCA2a, which initiated calcium overload. In addition, ischemic injury-triggered [Ca]c overload and subsequent [Ca]m overload led to mPTP opening and ΔΨm dissipation compared with the control. Furthermore, ablation of Mfn2 alleviated SERCA2a-induced mitochondrial calcium overload and subsequent mito-apoptosis in the context of CMEC hypoxic injury. , compared with that in wild-type mice, the myocardial infarct size in Mfn2 mice was significantly decreased. In addition, the findings revealed that Mfn2 mice had better heart contractile function, decreased myocardial infarction indicators, and improved mitochondrial morphology. Taken together, the results of our study suggested that SERCA2a-dependent [Ca]c overload led to mitochondrial dysfunction and activation of Mfn2-mediated [Ca]m overload. Overexpression of SERCA2a or ablation of Mfn2 expression mitigated mitochondrial morphological and functional damage by modifying the SERCA2a/Ca-Mfn2 pathway. Overall, these pathways are promising therapeutic targets for acute cardiac microvascular ischemic injury.
我们之前的研究表明,在心脏过度氧化损伤的情况下,2a型肌浆网/内质网钙ATP酶(SERCA2a)在心脏微血管内皮细胞(CMECs)中会发生转录后氧化修饰。然而,SERCA2a失活是否会导致线粒体稳态中的胞质钙失衡尚不清楚。线粒体融合蛋白2(Mfn2)是一种众所周知的参与内质网(ER)/线粒体钙连接和线粒体质量调节的重要蛋白质。因此,我们研究的目的是阐明在心脏微血管缺血损伤条件下,SERCA2a通过Mfn2连接介导线粒体钙超载的具体机制以及心脏的存活率。从小鼠中提取的CMECs进行6小时的缺氧损伤以模拟缺血性心脏损伤。通过结扎左前降支对C57-WT和Mfn2基因敲除小鼠进行1小时的缺血操作,以建立心脏缺血损伤模型。采用TTC染色、免疫组织化学和超声心动图来评估心肌梗死面积、微血管损伤和心脏功能。缺血损伤诱导了SERCA2a不可逆的氧化修饰,包括半胱氨酸674的磺酰化和酪氨酸294/295的硝化,以及SERCA2a失活,从而引发钙超载。此外,与对照组相比,缺血损伤引发的[Ca]c超载和随后的[Ca]m超载导致线粒体通透性转换孔(mPTP)开放和线粒体膜电位(ΔΨm)耗散。此外,在CMEC缺氧损伤的情况下,敲除Mfn2可减轻SERCA2a诱导的线粒体钙超载和随后的线粒体凋亡。与野生型小鼠相比,Mfn2基因敲除小鼠的心肌梗死面积显著减小。此外,研究结果显示,Mfn2基因敲除小鼠具有更好的心脏收缩功能,心肌梗死指标降低,线粒体形态得到改善。综上所述,我们的研究结果表明,SERCA2a依赖性的[Ca]c超载导致线粒体功能障碍并激活Mfn2介导的[Ca]m超载。SERCA2a的过表达或Mfn2表达的敲除通过改变SERCA2a/Ca-Mfn2途径减轻了线粒体形态和功能损伤。总体而言,这些途径是急性心脏微血管缺血损伤有前景的治疗靶点。