Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY 10021, USA.
Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY 10021, USA.
Brain Behav Immun. 2021 Jul;95:489-501. doi: 10.1016/j.bbi.2021.04.010. Epub 2021 Apr 17.
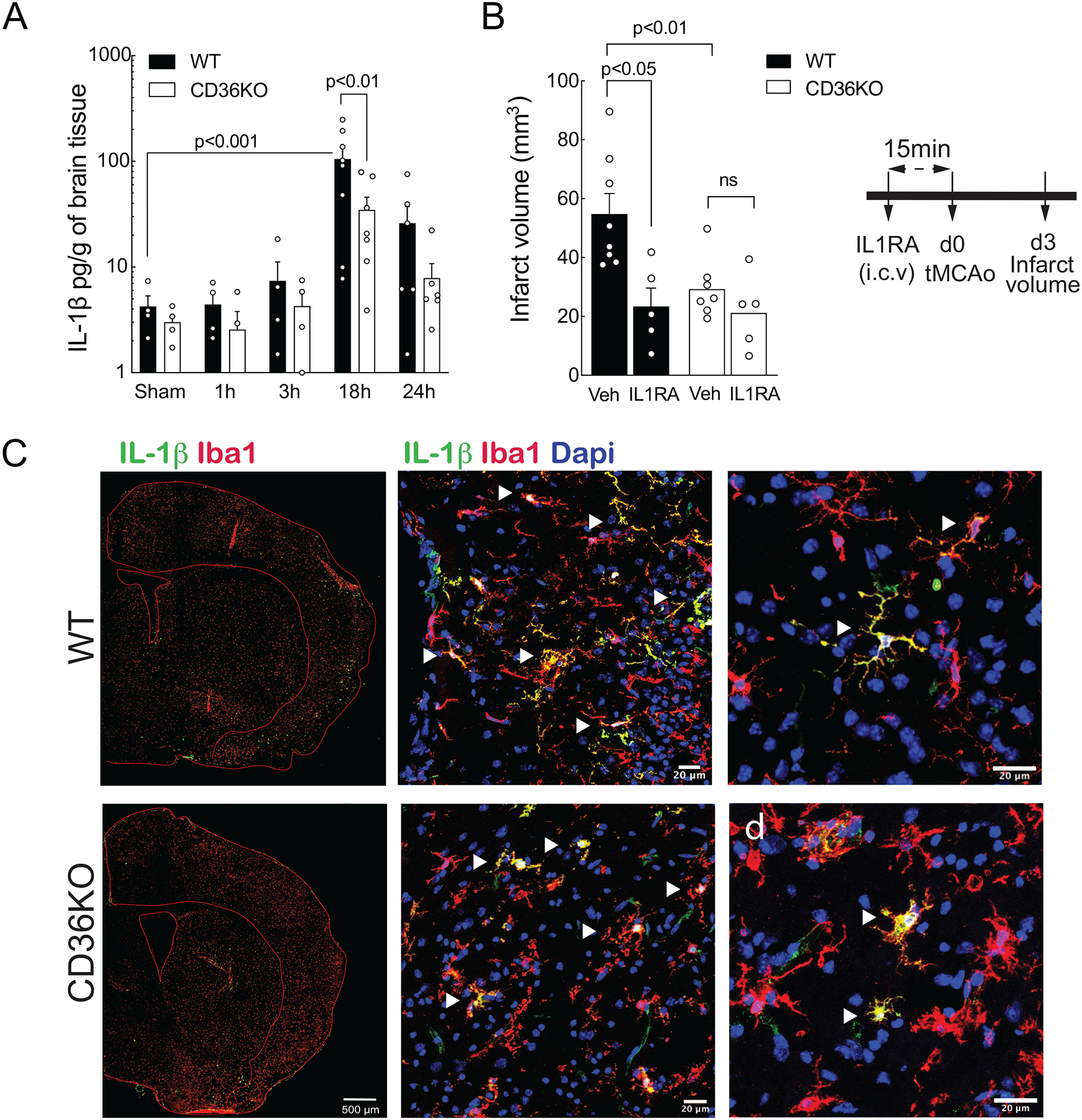
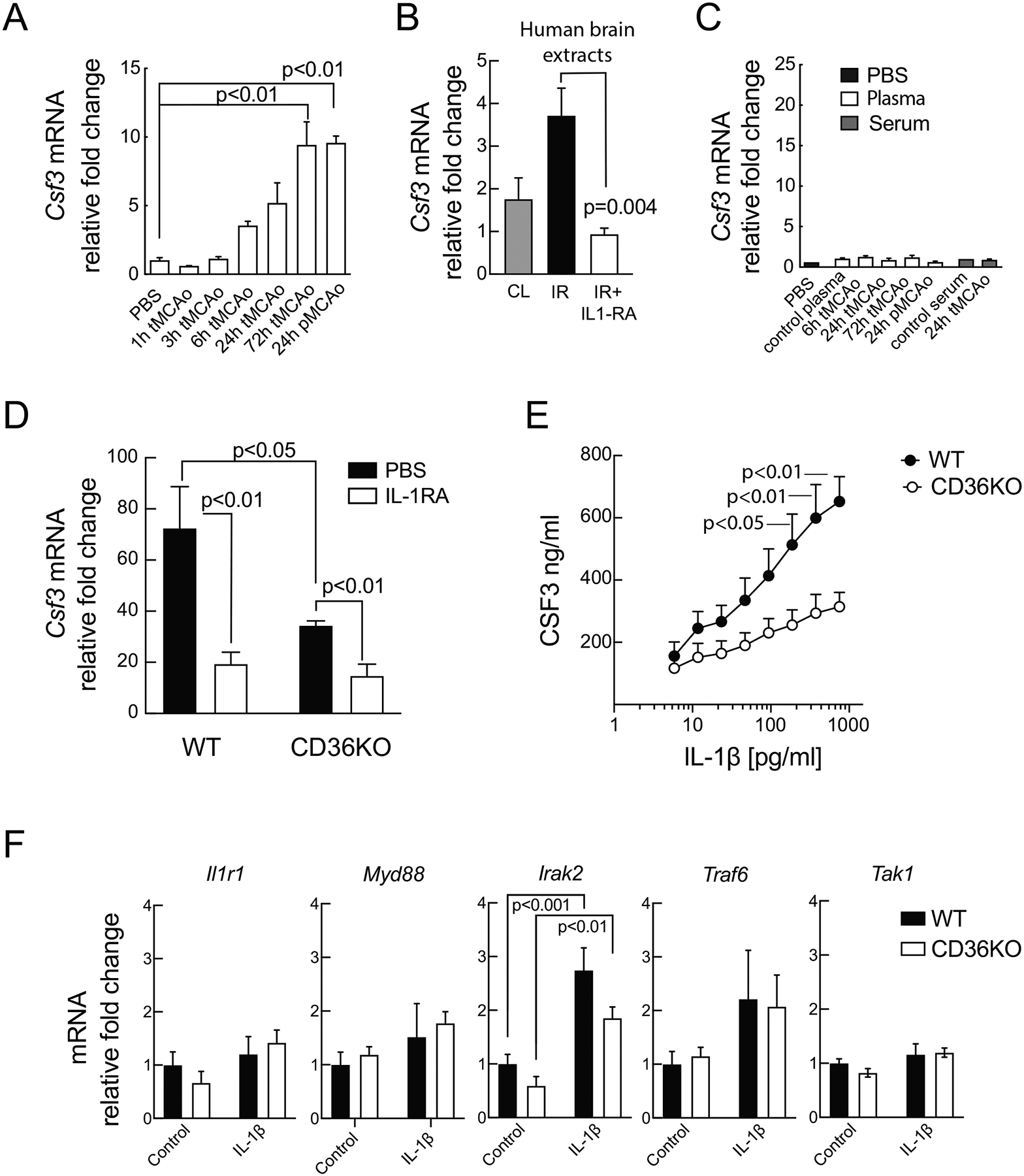
Cerebral ischemia is associated with an acute inflammatory response that contributes to the resulting injury. The innate immunity receptor CD36, expressed in microglia and endothelium, and the pro-inflammatory cytokine interleukin-1β (IL-1β) are involved in the mechanisms of ischemic injury. Since CD36 has been implicated in activation of the inflammasome, the main source of IL-1β, we investigated whether CD36 mediates brain injury through the inflammasome and IL-1β. We found that active caspase-1, a key inflammasome component, is decreased in microglia of CD36-deficient mice subjected to transient middle cerebral artery occlusion, an effect associated with a reduction in brain IL-1β. Conditional deletion of CD36 either in microglia or endothelium reduced ischemic injury in mice, attesting to the pathogenic involvement of CD36 in both cell types. Application of an ischemic brain extract to primary brain endothelial cell cultures from wild type (WT) mice induced IL-1β-dependent endothelial activation, reflected by increases in the cytokine colony stimulating factor-3, a response markedly attenuated in CD36-deficient endothelia. Similarly, the increase in colony stimulating factor-3 induced by recombinant IL-1β was attenuated in CD36-deficient compared to WT endothelia. We conclude that microglial CD36 is a key determinant of post-ischemic IL-1β production by regulating caspase-1 activity, whereas endothelial CD36 is required for the full expression of the endothelial activation induced by IL-1β. The data identify microglial and endothelial CD36 as critical upstream components of the acute inflammatory response to cerebral ischemia and viable putative therapeutic targets.
脑缺血与急性炎症反应有关,后者有助于导致损伤。先天免疫受体 CD36 在小胶质细胞和内皮细胞中表达,并参与缺血性损伤的机制。由于 CD36 已被牵连到炎性小体(IL-1β 的主要来源)的激活中,因此我们研究了 CD36 是否通过炎性小体和 IL-1β介导脑损伤。我们发现,在经历短暂性大脑中动脉闭塞的 CD36 缺陷型小鼠的小胶质细胞中,活性半胱氨酸蛋白酶-1(炎性小体的关键成分)减少,这与脑 IL-1β 的减少有关。在小胶质细胞或内皮细胞中条件性缺失 CD36 可减少小鼠的缺血性损伤,证明 CD36 在这两种细胞类型中均具有致病性。将缺血性脑提取物应用于来自野生型(WT)小鼠的原代脑内皮细胞培养物中,可诱导 IL-1β 依赖性内皮细胞激活,表现为细胞因子集落刺激因子-3 的增加,而在 CD36 缺陷型内皮细胞中,该反应明显减弱。同样,与 WT 内皮细胞相比,重组 IL-1β 诱导的集落刺激因子-3 增加在 CD36 缺陷型内皮细胞中减弱。我们得出的结论是,小胶质细胞 CD36 通过调节半胱氨酸蛋白酶-1 的活性成为缺血后 IL-1β 产生的关键决定因素,而内皮细胞 CD36 是 IL-1β 诱导的内皮激活完全表达所必需的。这些数据确定了小胶质细胞和内皮细胞 CD36 是脑缺血后急性炎症反应的关键上游组成部分,也是可行的潜在治疗靶点。