Bioinformatics, Institute for Biochemistry and Biology, University of Potsdam, 14476, Potsdam, Germany.
Systems Biology and Mathematical Modeling, Max Planck Institute of Molecular Plant Physiology, 14476, Potsdam, Germany.
Sci Rep. 2021 Apr 20;11(1):8544. doi: 10.1038/s41598-021-87643-8.
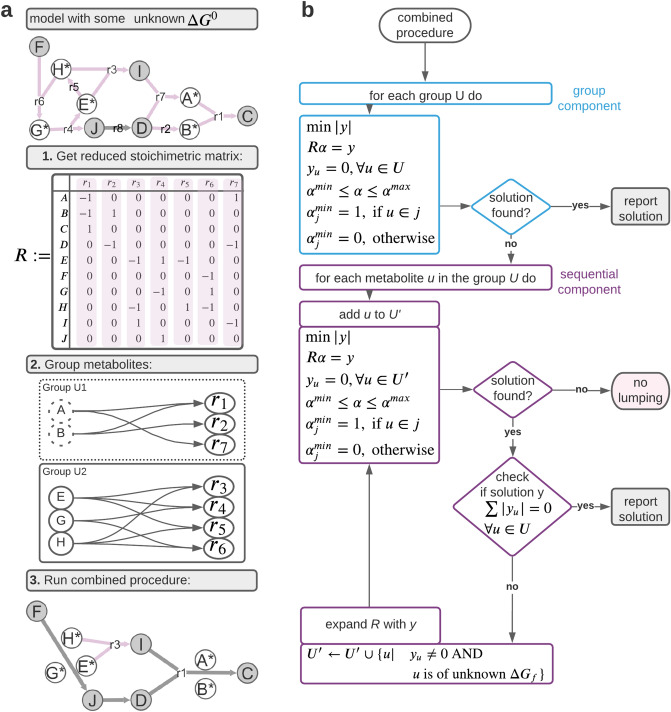
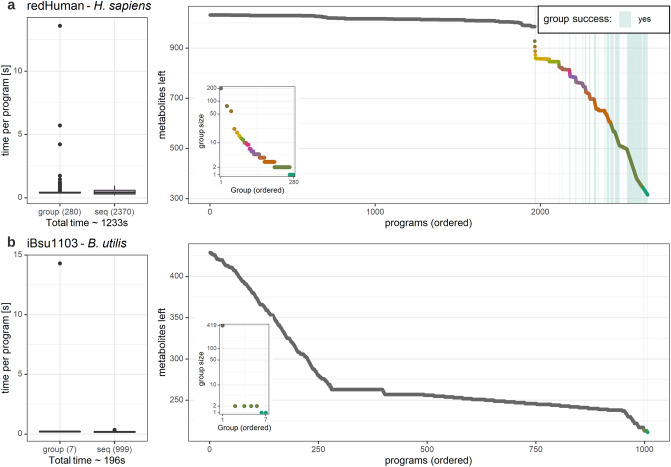
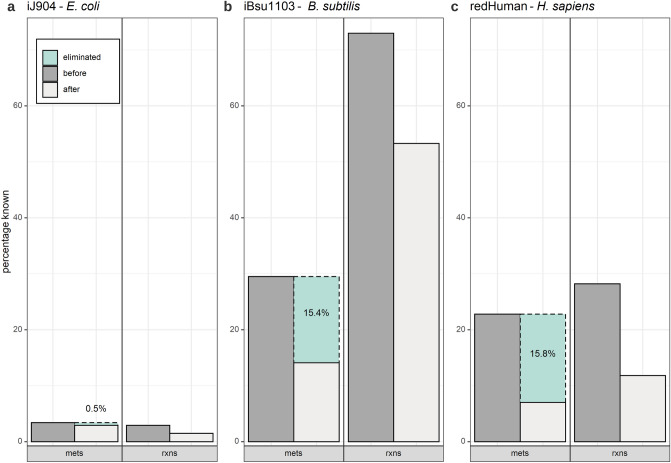
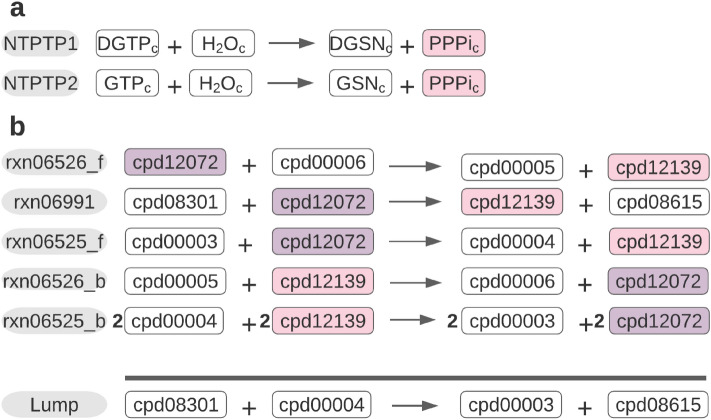
Thermodynamic metabolic flux analysis (TMFA) can narrow down the space of steady-state flux distributions, but requires knowledge of the standard Gibbs free energy for the modelled reactions. The latter are often not available due to unknown Gibbs free energy change of formation [Formula: see text], of metabolites. To optimize the usage of data on thermodynamics in constraining a model, reaction lumping has been proposed to eliminate metabolites with unknown [Formula: see text]. However, the lumping procedure has not been formalized nor implemented for systematic identification of lumped reactions. Here, we propose, implement, and test a combined procedure for reaction lumping, applicable to genome-scale metabolic models. It is based on identification of groups of metabolites with unknown [Formula: see text] whose elimination can be conducted independently of the others via: (1) group implementation, aiming to eliminate an entire such group, and, if this is infeasible, (2) a sequential implementation to ensure that a maximal number of metabolites with unknown [Formula: see text] are eliminated. Our comparative analysis with genome-scale metabolic models of Escherichia coli, Bacillus subtilis, and Homo sapiens shows that the combined procedure provides an efficient means for systematic identification of lumped reactions. We also demonstrate that TMFA applied to models with reactions lumped according to the proposed procedure lead to more precise predictions in comparison to the original models. The provided implementation thus ensures the reproducibility of the findings and their application with standard TMFA.
热力学代谢通量分析(TMFA)可以缩小稳态通量分布的空间,但需要对所建模反应的标准吉布斯自由能有所了解。由于代谢物形成吉布斯自由能变化[Formula: see text]未知,因此后者通常无法获得。为了优化在约束模型中使用热力学数据,已经提出了反应聚并来消除具有未知[Formula: see text]的代谢物。然而,该聚并过程尚未被形式化或实施,以系统地识别聚并反应。在这里,我们提出、实施并测试了一种适用于基因组规模代谢模型的组合反应聚并方法。它基于识别具有未知[Formula: see text]的代谢物组,通过以下方式可以独立于其他代谢物消除这些代谢物:(1)组实施,旨在消除整个这样的组,如果不可行,则(2)顺序实施,以确保消除具有未知[Formula: see text]的最大数量的代谢物。我们对大肠杆菌、枯草芽孢杆菌和人类的基因组规模代谢模型的比较分析表明,组合方法为系统识别聚并反应提供了一种有效的手段。我们还证明,与根据所提出的方法聚并反应的模型相比,应用 TMFA 可以更精确地预测。因此,提供的实现确保了发现的可重复性及其与标准 TMFA 的应用。