Institute of Pathology, University of Bern, Bern, Switzerland.
Graduate School for Cellular and Biomedical Sciences, University of Bern, Bern, Switzerland.
Sci Rep. 2021 Apr 27;11(1):9011. doi: 10.1038/s41598-021-87966-6.
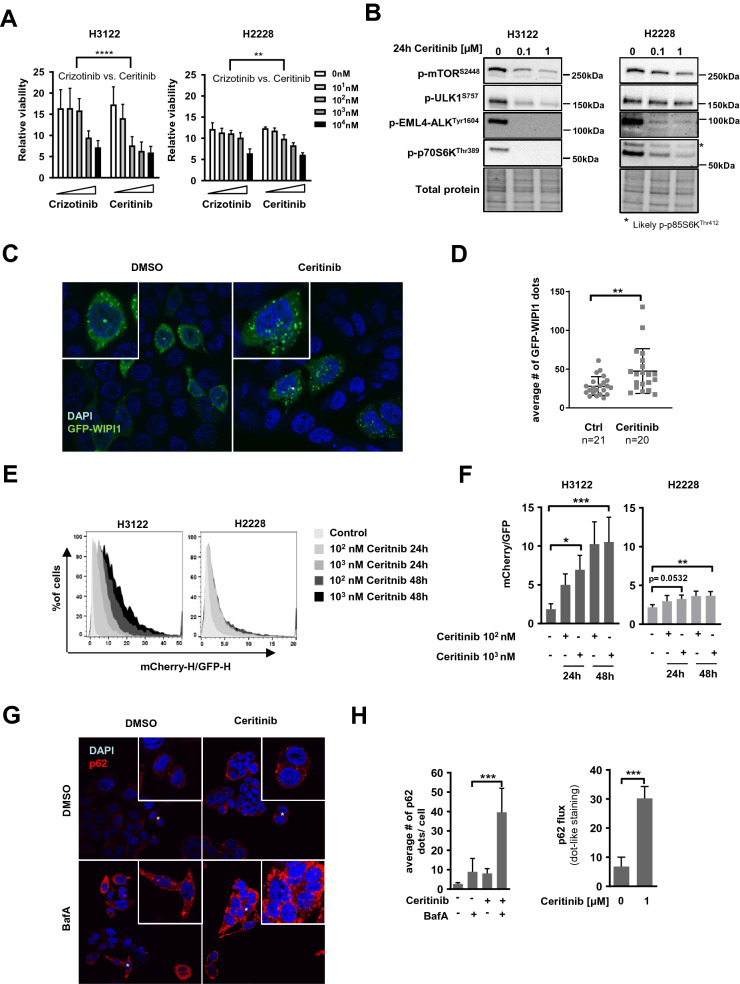
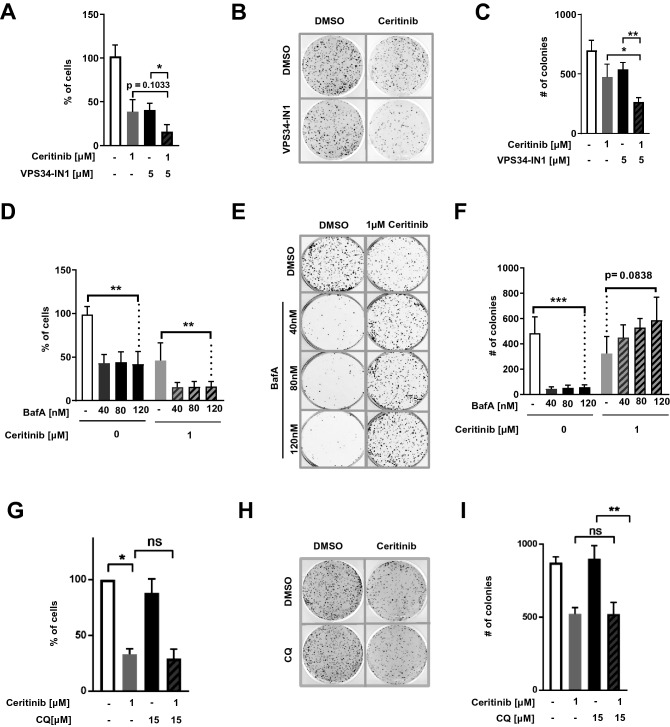
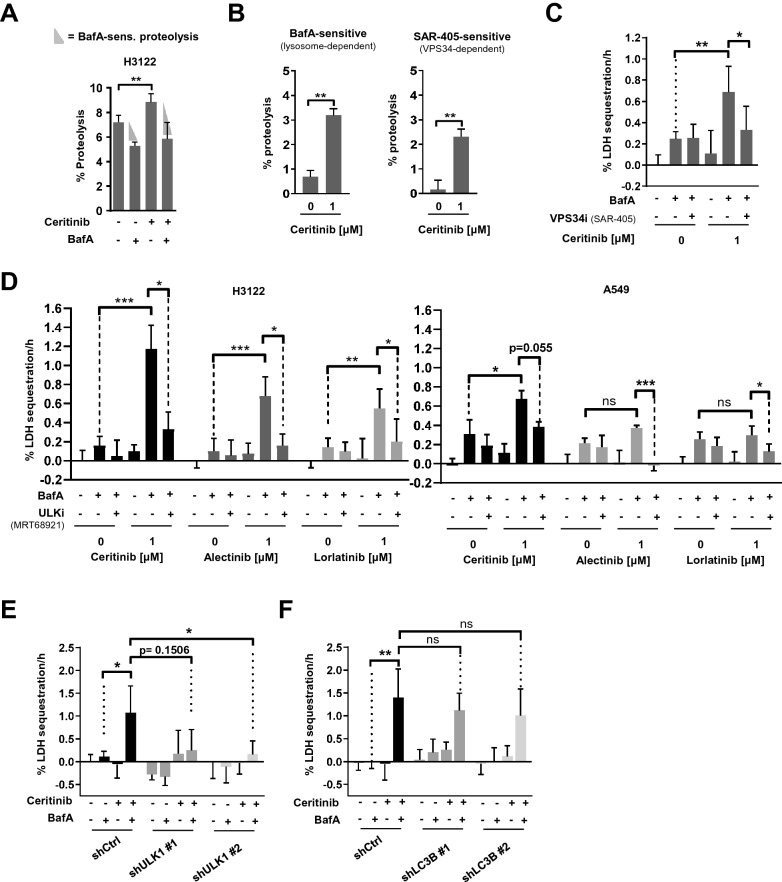
ALK inhibitors effectively target EML4-ALK positive non-small cell lung cancer, but their effects are hampered by treatment resistance. In the present study, we asked whether ALK inhibition affects autophagy, and whether this may influence treatment response. Whereas the impact of targeted therapies on autophagic activity previously have been assessed by surrogate marker proteins such as LC3B, we here thoroughly examined effects on functional autophagic activity, i.e. on the sequestration and degradation of autophagic cargo, in addition to autophagic markers. Interestingly, the ALK inhibitor Ceritinib decreased mTOR activity and increased GFP-WIPI1 dot formation in H3122 and H2228 EML4-ALK lung cancer cells, suggesting autophagy activation. Moreover, an mCherry-EGFP-LC3B based assay indicated elevated LC3B carrier flux upon ALK inhibition. In accordance, autophagic cargo sequestration and long-lived protein degradation significantly increased upon ALK inhibition. Intriguingly, autophagic cargo flux was dependent on VPS34 and ULK1, but not LC3B. Co-treating H3122 cells with Ceritinib and a VPS34 inhibitor or Bafilomycin A1 resulted in reduced cell numbers. Moreover, VPS34 inhibition reduced clonogenic recovery of Ceritinib-treated cells. In summary, our results indicate that ALK inhibition triggers LC3B-independent macroautophagic flux in EML4-ALK cells to support cancer cell survival and clonogenic growth.
ALK 抑制剂能有效靶向治疗 EML4-ALK 阳性非小细胞肺癌,但它们的疗效受到耐药性的影响。在本研究中,我们探讨了 ALK 抑制是否会影响自噬,以及这是否会影响治疗反应。先前已有研究通过 LC3B 等替代标志物蛋白来评估靶向治疗对自噬活性的影响,但我们在此更全面地研究了其对功能性自噬活性(即自噬货物的隔离和降解)的影响,以及自噬标志物的影响。有趣的是,ALK 抑制剂色瑞替尼降低了 H3122 和 H2228 EML4-ALK 肺癌细胞中的 mTOR 活性,并增加了 GFP-WIPI1 斑点形成,表明自噬被激活。此外,基于 mCherry-EGFP-LC3B 的测定表明,ALK 抑制后 LC3B 载体通量增加。相应地,自噬货物的隔离和长寿命蛋白的降解在 ALK 抑制后显著增加。有趣的是,自噬货物的通量依赖于 VPS34 和 ULK1,而不依赖于 LC3B。在 H3122 细胞中,色瑞替尼与 VPS34 抑制剂或巴弗洛霉素 A1 共同处理会导致细胞数量减少。此外,VPS34 抑制会降低色瑞替尼处理细胞的集落形成恢复能力。综上所述,我们的研究结果表明,ALK 抑制在 EML4-ALK 细胞中触发了 LC3B 非依赖性巨自噬通量,以支持癌细胞的存活和集落生长。