Tang Rong, Liu Xiaomeng, Wang Wei, Hua Jie, Xu Jin, Liang Chen, Meng Qingcai, Liu Jiang, Zhang Bo, Yu Xianjun, Shi Si
Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, Shanghai, China.
Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China.
Front Oncol. 2021 Apr 12;11:643465. doi: 10.3389/fonc.2021.643465. eCollection 2021.
Cancer stem cells (CSCs) are widely thought to contribute to the dismal prognosis of pancreatic ductal adenocarcinoma (PDAC). CSCs share biological features with adult stem cells, such as longevity, self-renewal capacity, differentiation, drug resistance, and the requirement for a niche; these features play a decisive role in cancer progression. A prominent characteristic of PDAC is metabolic reprogramming, which provides sufficient nutrients to support rapid tumor cell growth. However, whether PDAC stemness is correlated with metabolic reprogramming remains unknown.
RNA sequencing data of PDAC, including read counts and fragments per kilobase of transcript per million mapped reads (FPKM), were collected from The Cancer Genome Atlas-Pancreatic Adenocarcinoma (TCGA-PAAD) database. Single-sample gene set enrichment analysis (GSEA) was used to calculate the relative activities of metabolic pathways in each PDAC sample. Quantitative real-time PCR was performed to validate the expression levels of genes of interest.
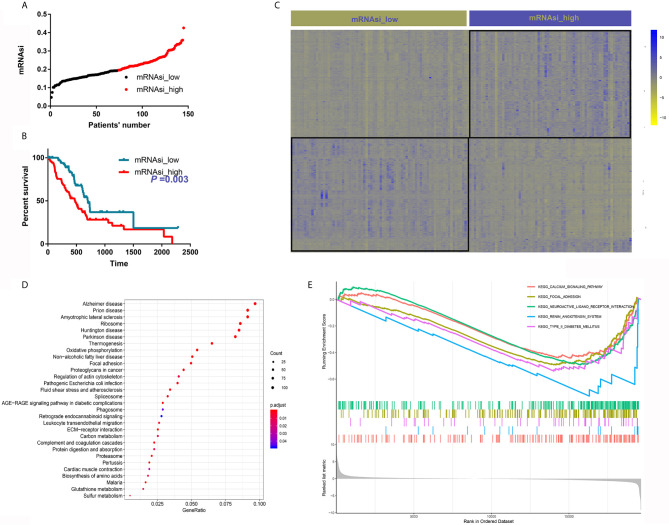
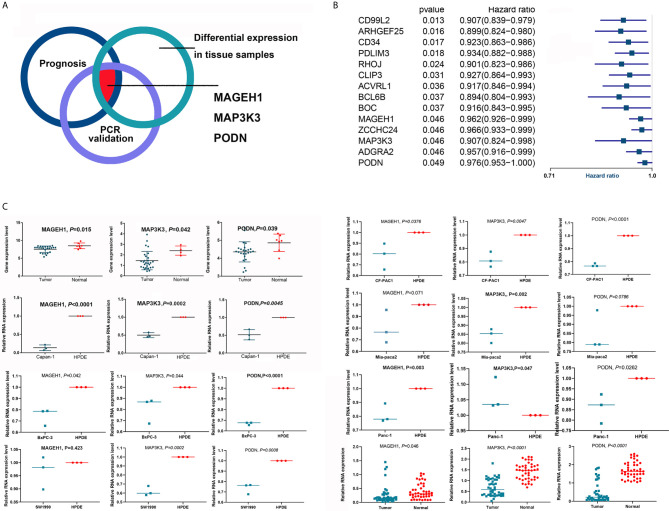
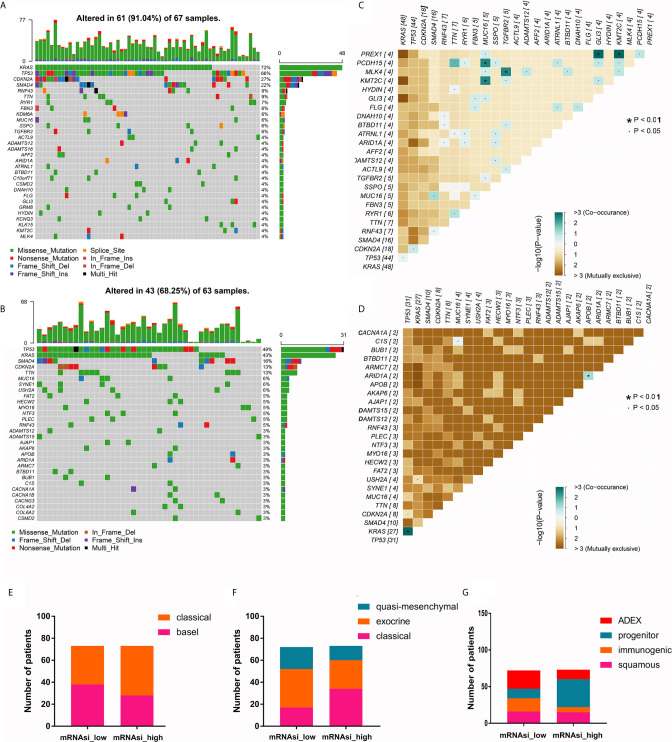
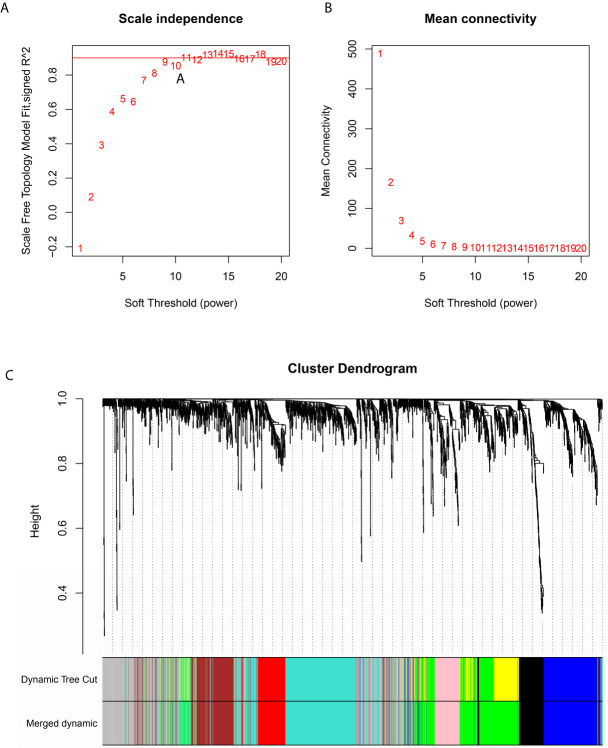
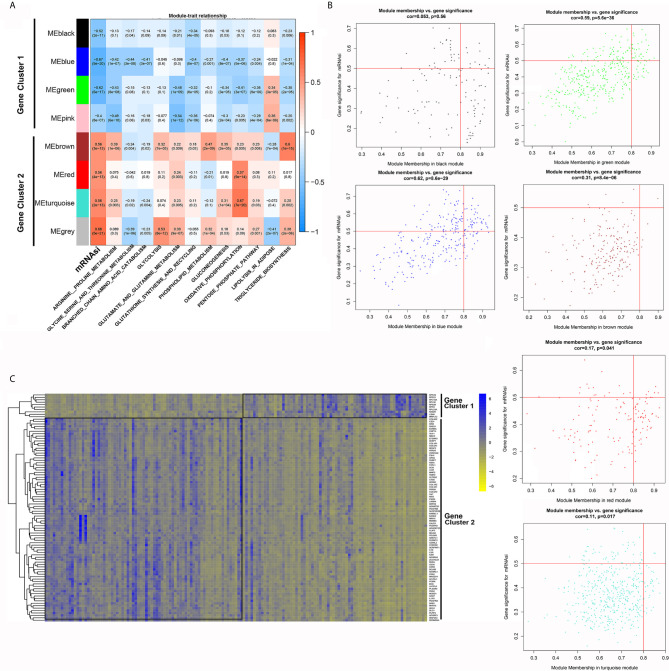
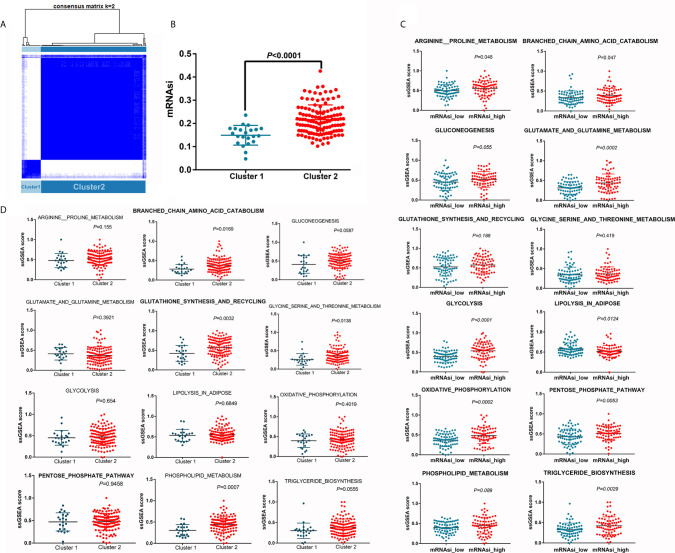
The overall survival (OS) of patients with high mRNA expression-based stemness index (mRNAsi) values was significantly worse than that of their counterparts with low mRNAsi values ( = 0.003). This survival disadvantage was independent of baseline clinical characteristics. Gene ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and GSEA showed that the differentially expressed genes between patients with high and low mRNAsi values were mainly enriched in oncogenic and metabolic pathways. Weighted gene coexpression network analysis (WGCNA) revealed 8 independent gene modules that were significantly associated with mRNAsi and 12 metabolic pathways. Unsupervised clustering based on the key genes in each module identified two PDAC subgroups characterized by different mRNAsi values and metabolic activities. Univariate Cox regression analysis identified 14 genes beneficial to OS from 95 key genes selected from the eight independent gene modules from WGCNA. Among them, MAGEH1, MAP3K3, and PODN were downregulated in both pancreatic tissues and cell lines.
The present study showed that PDAC samples with high mRNAsi values exhibited aberrant activation of multiple metabolic pathways, and the patients from whom these samples were obtained had a poor prognosis. Future studies are expected to investigate the underlying mechanism based on the crosstalk between PDAC stemness and metabolic rewiring.
癌症干细胞(CSCs)被广泛认为是导致胰腺导管腺癌(PDAC)预后不良的原因。CSCs与成体干细胞具有共同的生物学特性,如寿命长、自我更新能力、分化、耐药性以及对特定微环境的需求;这些特性在癌症进展中起决定性作用。PDAC的一个突出特征是代谢重编程,它为肿瘤细胞的快速生长提供了充足的营养。然而,PDAC的干性是否与代谢重编程相关仍不清楚。
从癌症基因组图谱-胰腺腺癌(TCGA-PAAD)数据库收集PDAC的RNA测序数据,包括读数计数和每百万映射读数中每千碱基转录本的片段数(FPKM)。使用单样本基因集富集分析(GSEA)计算每个PDAC样本中代谢途径的相对活性。进行定量实时PCR以验证感兴趣基因的表达水平。
基于mRNA的干性指数(mRNAsi)值高的患者的总生存期(OS)明显差于mRNAsi值低的患者(P = 0.003)。这种生存劣势与基线临床特征无关。基因本体(GO)分析、京都基因与基因组百科全书(KEGG)分析和GSEA表明,mRNAsi值高和低的患者之间差异表达的基因主要富集在致癌和代谢途径中。加权基因共表达网络分析(WGCNA)揭示了8个与mRNAsi和12条代谢途径显著相关的独立基因模块。基于每个模块中的关键基因进行无监督聚类,确定了两个以不同mRNAsi值和代谢活性为特征的PDAC亚组。单变量Cox回归分析从WGCNA的八个独立基因模块中选择的95个关键基因中确定了14个对OS有益的基因。其中,MAGEH1、MAP3K3和PODN在胰腺组织和细胞系中均下调。
本研究表明,mRNAsi值高的PDAC样本表现出多种代谢途径的异常激活,且获取这些样本的患者预后较差。未来的研究有望基于PDAC干性与代谢重塑之间的相互作用来研究其潜在机制。