Computational and Theoretical Chemistry Group (CATCO), Department of Chemistry, Southern Methodist University, 3215 Daniel Ave, Dallas, TX 75275-0314, USA.
Molecules. 2021 Apr 14;26(8):2268. doi: 10.3390/molecules26082268.
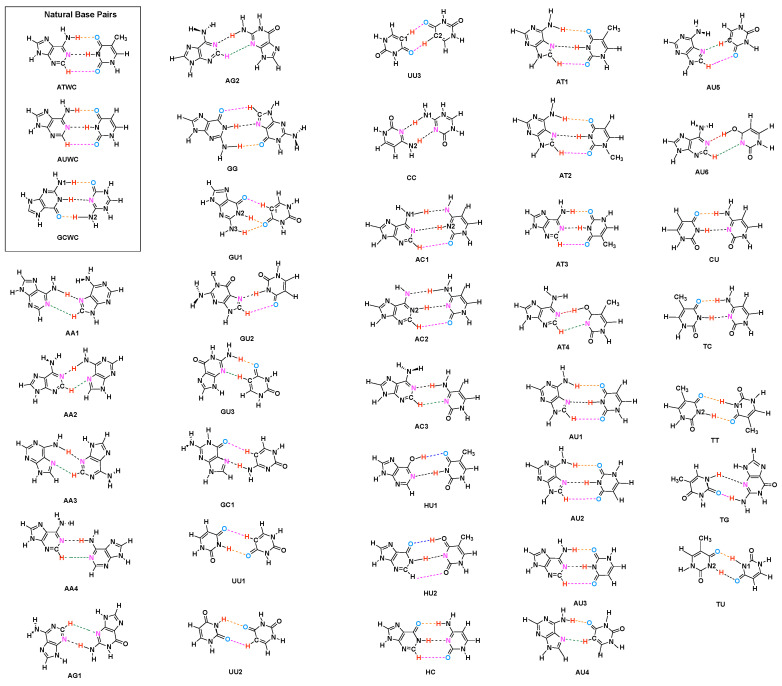
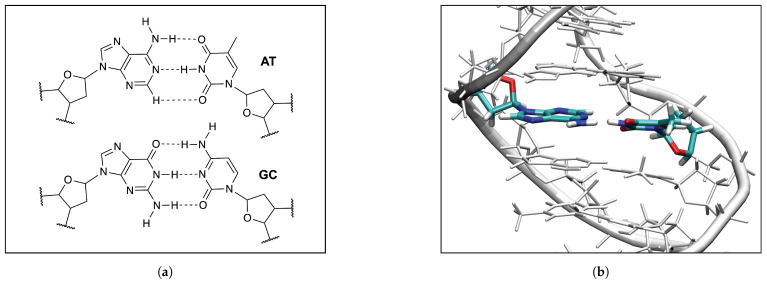
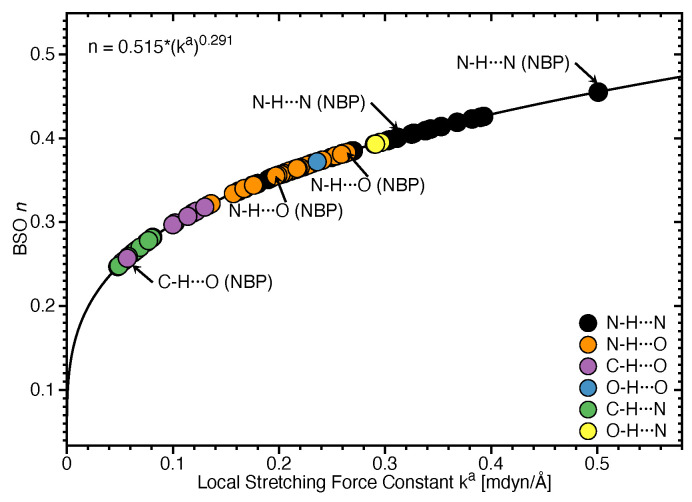
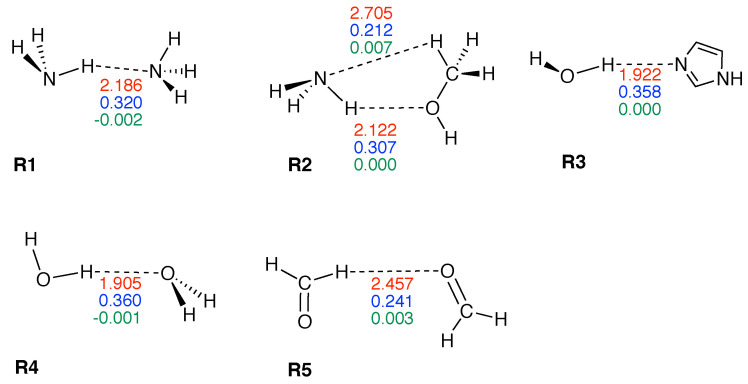
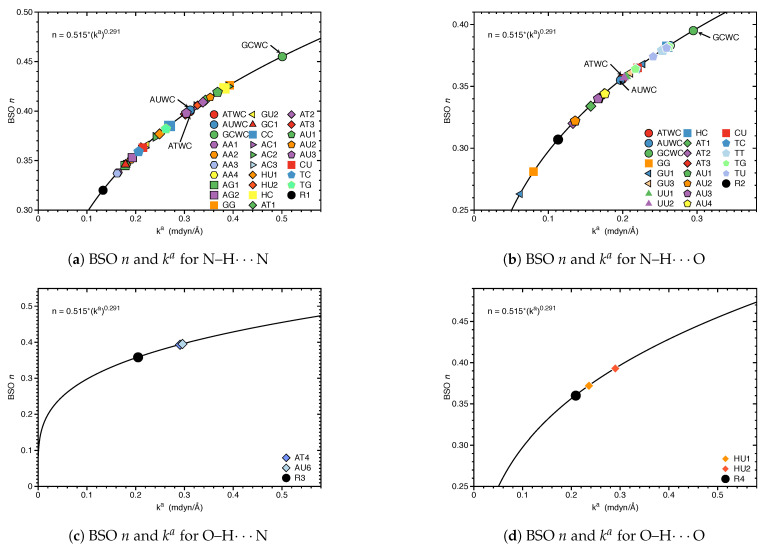
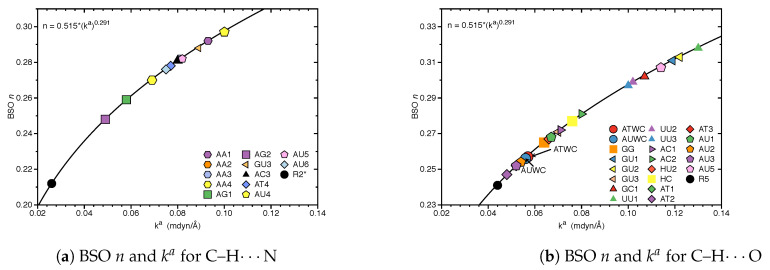
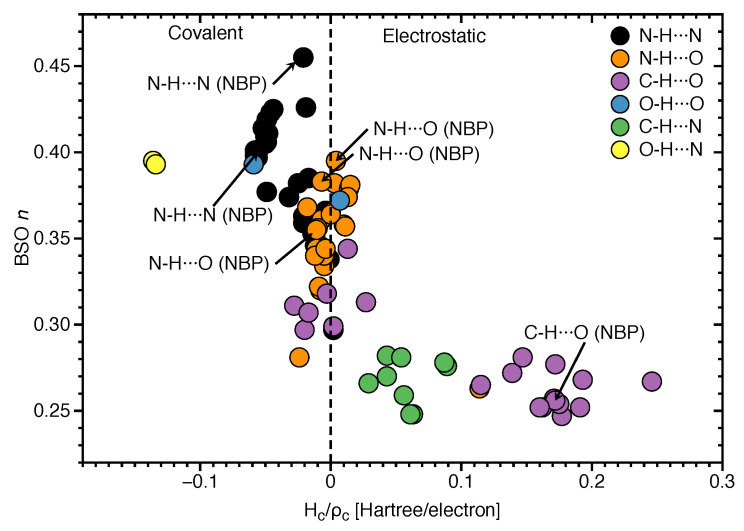
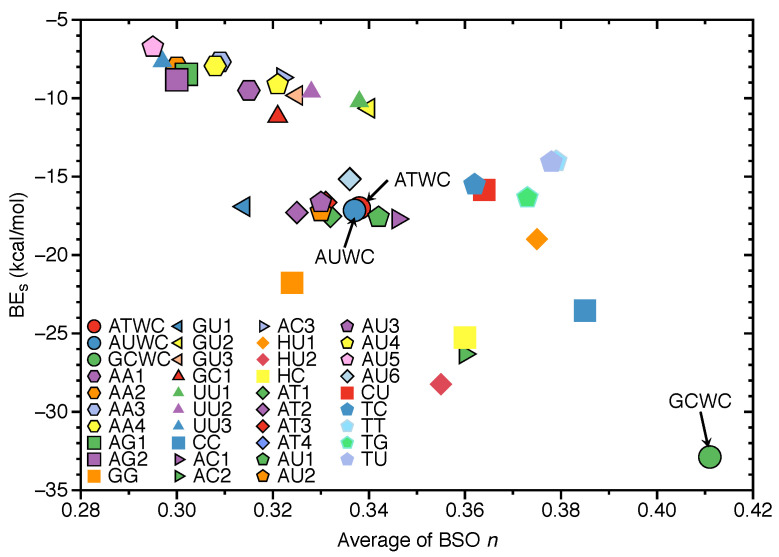
In this work hydrogen bonding in a diverse set of 36 unnatural and the three natural Watson Crick base pairs adenine (A)-thymine (T), adenine (A)-uracil (U) and guanine (G)-cytosine (C) was assessed utilizing local vibrational force constants derived from the local mode analysis, originally introduced by Konkoli and Cremer as a unique bond strength measure based on vibrational spectroscopy. The local mode analysis was complemented by the topological analysis of the electronic density and the natural bond orbital analysis. The most interesting findings of our study are that (i) hydrogen bonding in Watson Crick base pairs is not exceptionally strong and (ii) the N-H⋯N is the most favorable hydrogen bond in both unnatural and natural base pairs while O-H⋯N/O bonds are the less favorable in unnatural base pairs and not found at all in natural base pairs. In addition, the important role of non-classical C-H⋯N/O bonds for the stabilization of base pairs was revealed, especially the role of C-H⋯O bonds in Watson Crick base pairs. Hydrogen bonding in Watson Crick base pairs modeled in the DNA via a QM/MM approach showed that the DNA environment increases the strength of the central N-H⋯N bond and the C-H⋯O bonds, and at the same time decreases the strength of the N-H⋯O bond. However, the general trends observed in the gas phase calculations remain unchanged. The new methodology presented and tested in this work provides the bioengineering community with an efficient design tool to assess and predict the type and strength of hydrogen bonding in artificial base pairs.
在这项工作中,利用最初由 Konkoli 和 Cremer 引入的局部振动力常数,对 36 种不同的非天然和三种天然 Watson-Crick 碱基对(腺嘌呤(A)-胸腺嘧啶(T)、腺嘌呤(A)-尿嘧啶(U)和鸟嘌呤(G)-胞嘧啶(C)中的氢键进行了评估。局部模式分析得到了局部模式分析的补充,包括电子密度的拓扑分析和自然键轨道分析。我们研究的最有趣的发现是:(i)Watson-Crick 碱基对中的氢键并不特别强,(ii)在非天然和天然碱基对中,N-H⋯N 是最有利的氢键,而 O-H⋯N/O 键在非天然碱基对中不太有利,在天然碱基对中根本不存在。此外,还揭示了非经典 C-H⋯N/O 键对碱基对稳定的重要作用,特别是 C-H⋯O 键在 Watson-Crick 碱基对中的作用。通过 QM/MM 方法在 DNA 中模拟 Watson-Crick 碱基对的氢键表明,DNA 环境增加了中心 N-H⋯N 键和 C-H⋯O 键的强度,同时降低了 N-H⋯O 键的强度。然而,在气相计算中观察到的总体趋势保持不变。本工作提出并测试的新方法为生物工程界提供了一种有效的设计工具,用于评估和预测人工碱基对中氢键的类型和强度。