Kazantsev Daniil, Duman Ramona, Wagner Armin, Mykhaylyk Vitaliy, Wanelik Kazimir, Basham Mark, Wadeson Nicola
Diamond Light Source Ltd, Diamond House, Harwell Science and Innovation Campus, Fermi Avenue, Didcot OX11 0DE United Kingdom.
J Synchrotron Radiat. 2021 May 1;28(Pt 3):889-901. doi: 10.1107/S1600577521003453. Epub 2021 Apr 20.
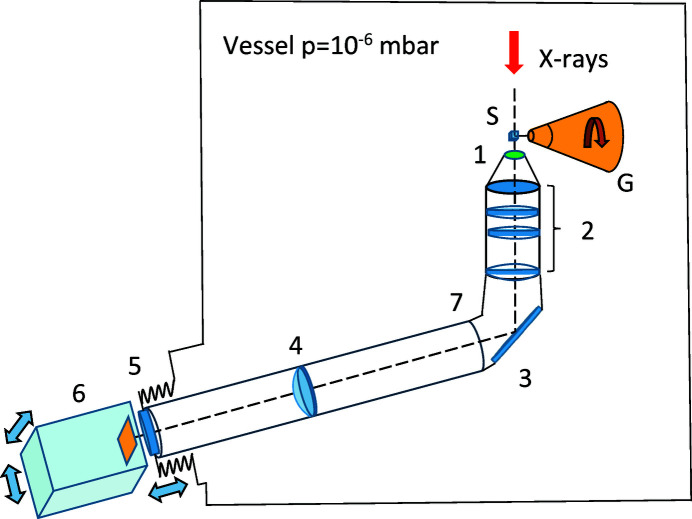
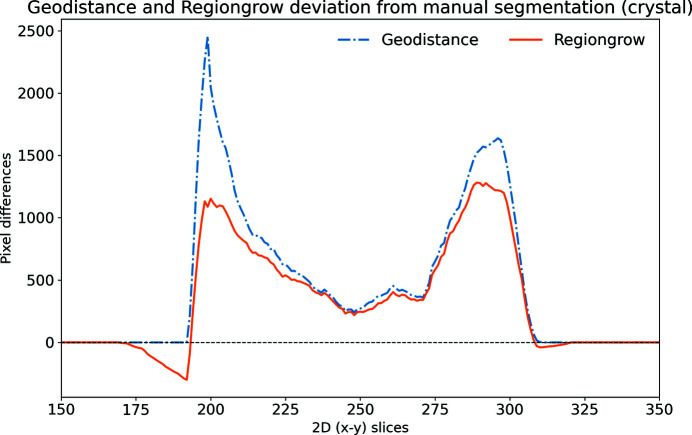
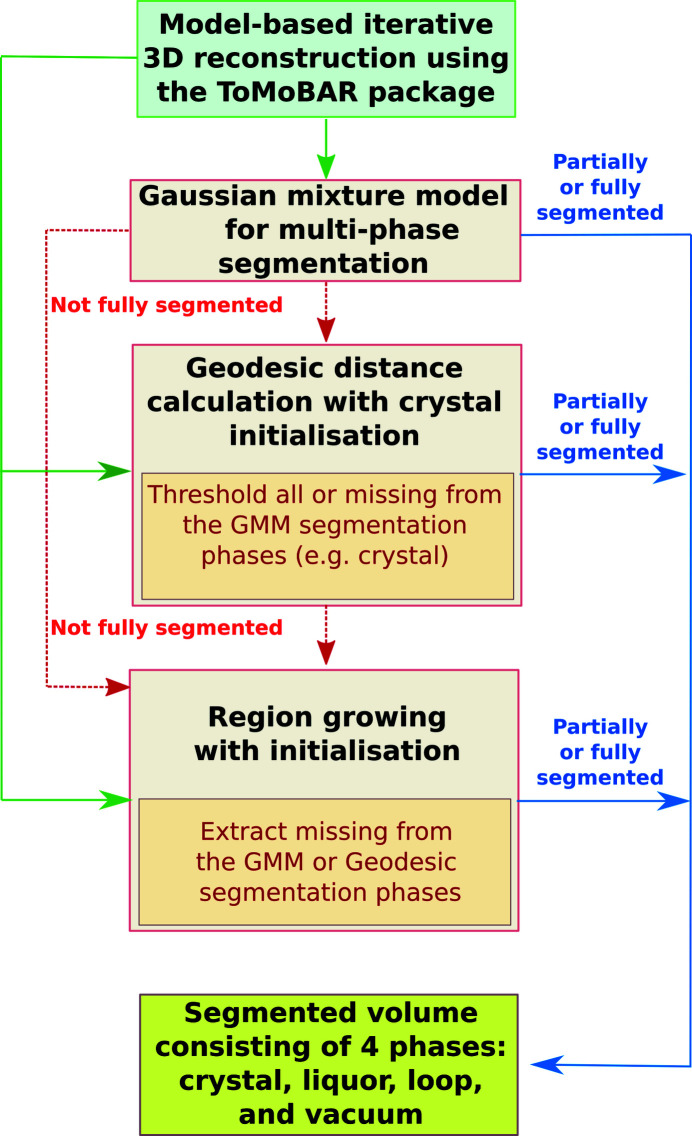
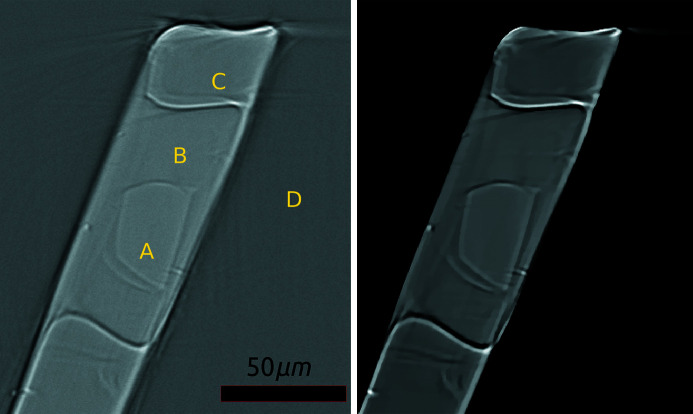
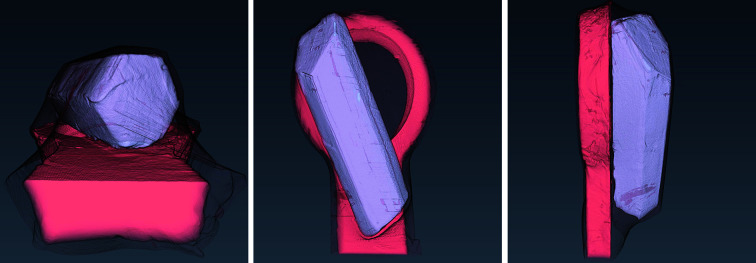
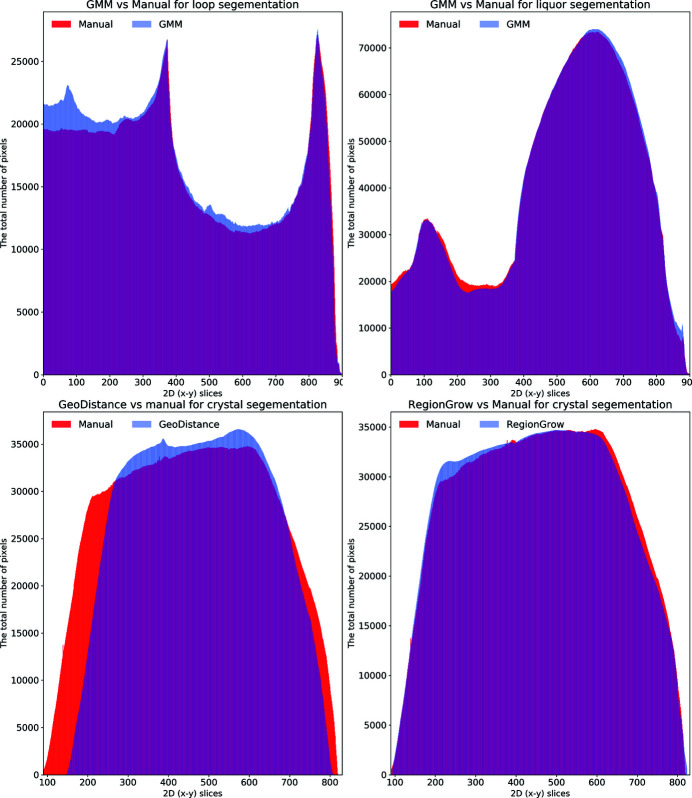
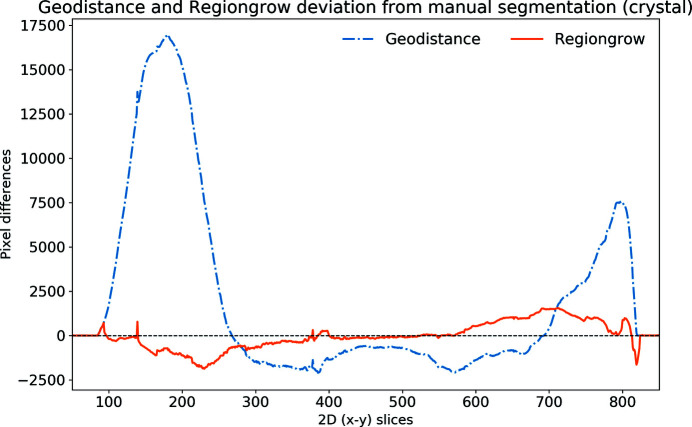
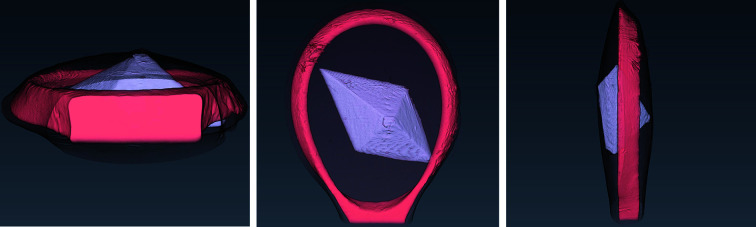
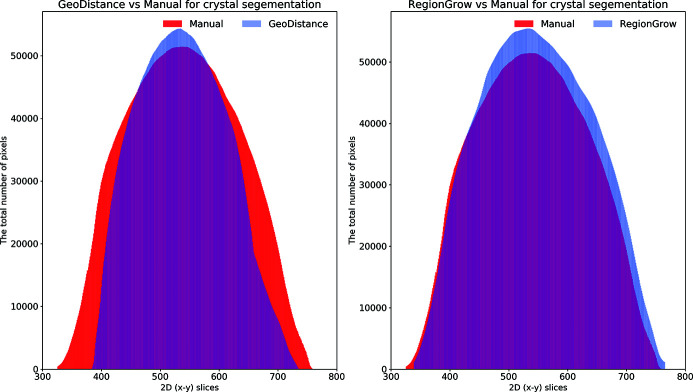
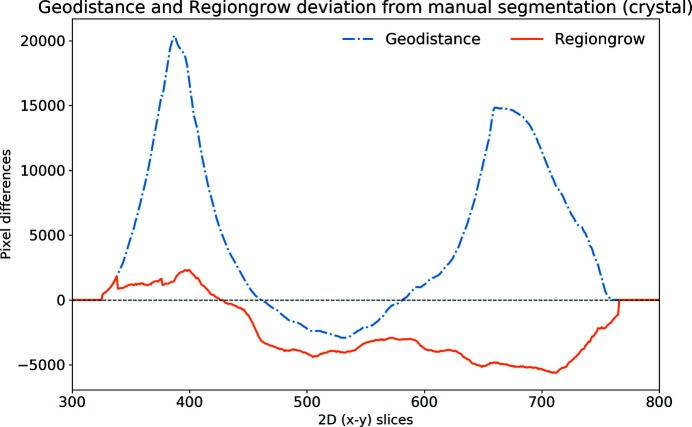
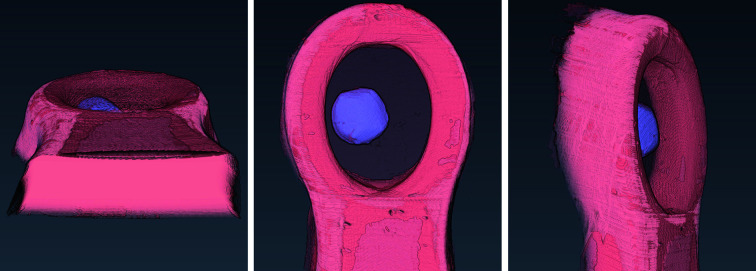
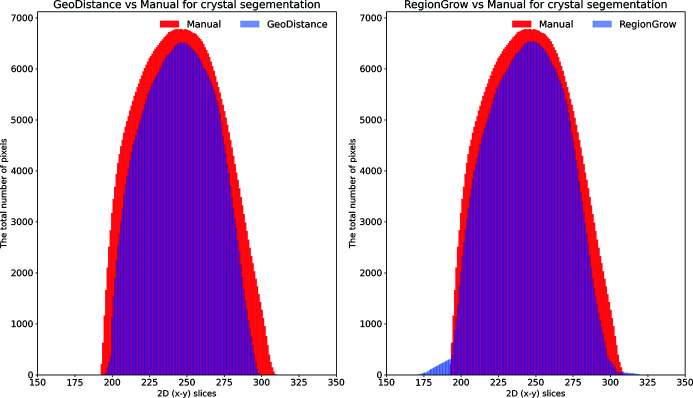
In this paper a practical solution for the reconstruction and segmentation of low-contrast X-ray tomographic data of protein crystals from the long-wavelength macromolecular crystallography beamline I23 at Diamond Light Source is provided. The resulting segmented data will provide the path lengths through both diffracting and non-diffracting materials as basis for analytical absorption corrections for X-ray diffraction data taken in the same sample environment ahead of the tomography experiment. X-ray tomography data from protein crystals can be difficult to analyse due to very low or absent contrast between the different materials: the crystal, the sample holder and the surrounding mother liquor. The proposed data processing pipeline consists of two major sequential operations: model-based iterative reconstruction to improve contrast and minimize the influence of noise and artefacts, followed by segmentation. The segmentation aims to partition the reconstructed data into four phases: the crystal, mother liquor, loop and vacuum. In this study three different semi-automated segmentation methods are experimented with by using Gaussian mixture models, geodesic distance thresholding and a novel morphological method, RegionGrow, implemented specifically for the task. The complete reconstruction-segmentation pipeline is integrated into the MPI-based data analysis and reconstruction framework Savu, which is used to reduce computation time through parallelization across a computing cluster and makes the developed methods easily accessible.
本文提供了一种实用的解决方案,用于对来自钻石光源长波长大分子晶体学光束线I23的蛋白质晶体低对比度X射线断层扫描数据进行重建和分割。所得的分割数据将提供穿过衍射和非衍射材料的路径长度,作为在断层扫描实验之前在相同样品环境中采集的X射线衍射数据的分析吸收校正的基础。由于不同材料(晶体、样品架和周围的母液)之间的对比度非常低或不存在,蛋白质晶体的X射线断层扫描数据可能难以分析。所提出的数据处理流程包括两个主要的顺序操作:基于模型的迭代重建,以提高对比度并最小化噪声和伪影的影响,随后进行分割。分割旨在将重建数据划分为四个阶段:晶体、母液、环和真空。在本研究中,通过使用高斯混合模型、测地距离阈值化和专门为该任务实现的一种新颖的形态学方法RegionGrow,对三种不同的半自动分割方法进行了试验。完整的重建 - 分割流程被集成到基于MPI的数据分析和重建框架Savu中,该框架用于通过跨计算集群并行化来减少计算时间,并使所开发的方法易于使用。