Université de Paris, IAME, UMR 1137, INSERM, F-75018, Paris, France.
LABGeM, Génomique Métabolique, Genoscope, Institut François Jacob, CEA, CNRS, Université Paris-Saclay, Evry, France.
Genome Med. 2021 May 5;13(1):77. doi: 10.1186/s13073-021-00892-0.
Escherichia coli is the leading cause of bloodstream infections, associated with a significant mortality. Recent genomic analyses revealed that few clonal lineages are involved in bloodstream infections and captured the emergence of some of them. However, data on within sequence type (ST) population genetic structure evolution are rare.
We compared whole genome sequences of 912 E. coli isolates responsible for bloodstream infections from two multicenter clinical trials that were conducted in the Paris area, France, 12 years apart, in teaching hospitals belonging to the same institution ("Assistance Publique-Hôpitaux de Paris"). We analyzed the strains at different levels of granularity, i.e., the phylogroup, the ST complex (STc), and the within STc clone taking into consideration the evolutionary history, the resistance, and virulence gene content as well as the antigenic diversity of the strains.
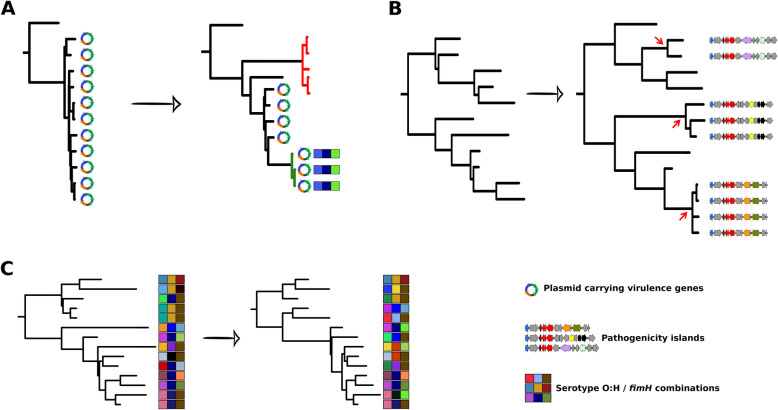
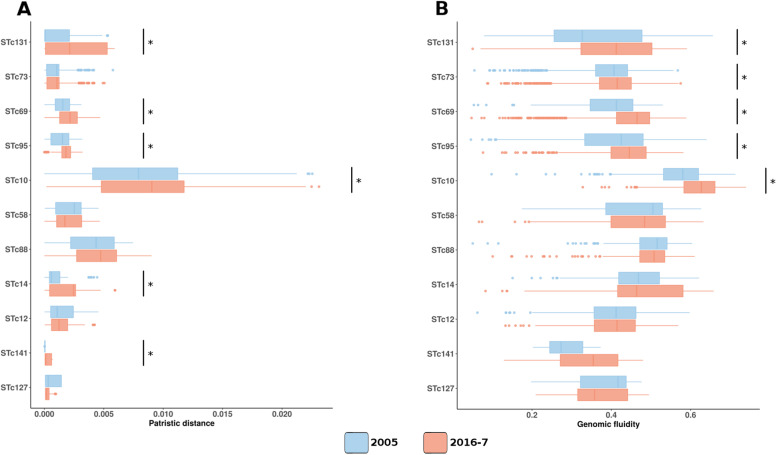
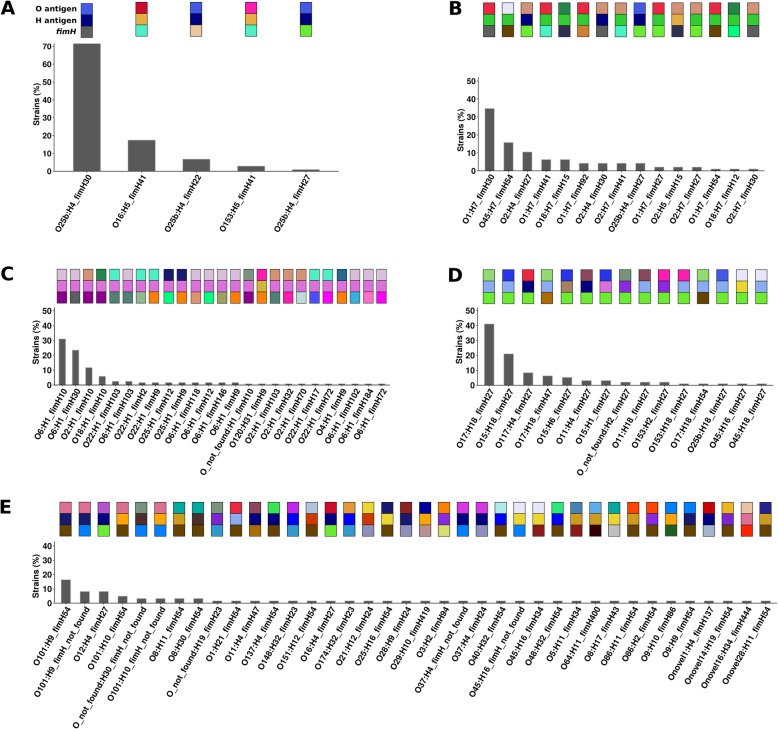
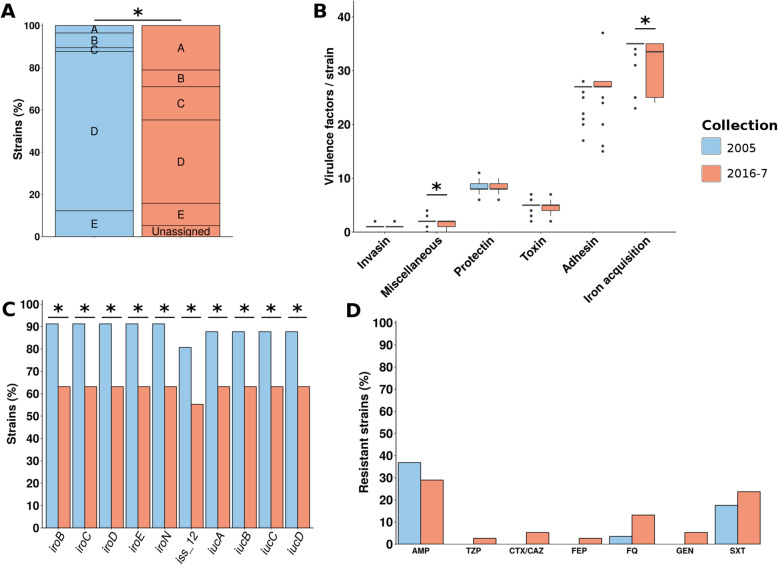
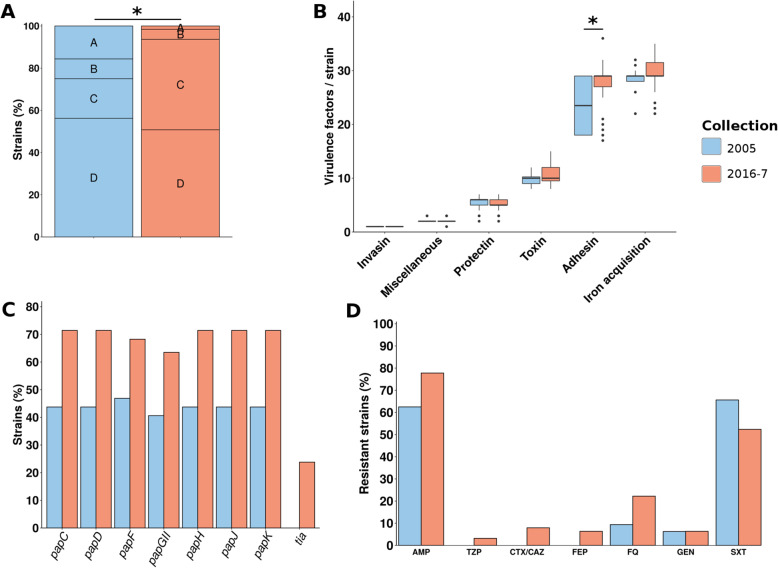
We found a mix of stability and changes overtime, depending on the level of comparison. Overall, we observed an increase in antibiotic resistance associated to a restricted number of genetic determinants and in strain plasmidic content, whereas phylogroup distribution and virulence gene content remained constant. Focusing on STcs highlighted the pauci-clonality of the populations, with only 11 STcs responsible for more than 73% of the cases, dominated by five STcs (STc73, STc131, STc95, STc69, STc10). However, some STcs underwent dramatic variations, such as the global pandemic STc131, which replaced the previously predominant STc95. Moreover, within STc131, 95 and 69 genomic diversity analysis revealed a highly dynamic pattern, with reshuffling of the population linked to clonal replacement sometimes coupled with independent acquisitions of virulence factors such as the pap gene cluster bearing a papGII allele located on various pathogenicity islands. Additionally, STc10 exhibited huge antigenic diversity evidenced by numerous O:H serotype/fimH allele combinations, whichever the year of isolation.
Altogether, these data suggest that the bloodstream niche is occupied by a wide but specific phylogenetic diversity and that highly specialized extra-intestinal clones undergo frequent turnover at the within ST level. Additional worldwide epidemiological studies overtime are needed in different geographical and ecological contexts to assess how generalizable these data are.
大肠杆菌是血流感染的主要原因,与较高的死亡率相关。最近的基因组分析显示,少数克隆谱系与血流感染有关,并捕捉到了其中一些谱系的出现。然而,关于序列型(ST)内种群遗传结构演变的数据很少。
我们比较了在法国巴黎地区两个多中心临床试验中分离的 912 株负责血流感染的大肠杆菌全基因组序列,两次试验相隔 12 年,这些菌株均来自同一机构的教学医院(“巴黎公立医院集团”)。我们在不同的粒度水平上分析了这些菌株,即进化群、ST 复合物(STc)和 STc 内的克隆,同时考虑了菌株的进化历史、耐药性和毒力基因含量以及抗原多样性。
我们发现,根据比较的水平,随着时间的推移,稳定性和变化并存。总体而言,我们观察到抗生素耐药性的增加与有限数量的遗传决定因素和菌株质粒含量的增加有关,而进化群分布和毒力基因含量保持不变。关注 STc 突出了种群的低克隆性,只有 11 个 STc 负责超过 73%的病例,其中以五个 STc(STc73、STc131、STc95、STc69、STc10)为主导。然而,一些 STc 发生了巨大的变化,例如全球大流行的 STc131,它取代了以前占主导地位的 STc95。此外,在 STc131 内,对基因组多样性的分析显示出高度动态的模式,种群的重新洗牌与克隆替换有关,有时与独立获得毒力因子(如携带位于各种致病性岛的 papGII 等位基因的 pap 基因簇)有关。此外,STc10 表现出巨大的抗原多样性,这从其不同年份的分离株的 O:H 血清型/fimH 等位基因组合中可以看出。
总之,这些数据表明,血流感染的生态位由广泛但特定的进化多样性占据,而高度专业化的肠外克隆在 ST 内水平上经常发生更替。需要在不同的地理和生态环境中进行更多的全球流行病学研究,以评估这些数据的普遍性。