Saini Poorvi, Bandsode Viraj, Singh Anuradha, Mendem Suresh Kumar, Semmler Torsten, Alam Munirul, Ahmed Niyaz
Department of Biotechnology and Bioinformatics, Pathogen Biology Laboratory, University of Hyderabad, Hyderabad, Telangana State, India.
Robert Koch Institute, Berlin, Germany.
mBio. 2024 Mar 13;15(3):e0354523. doi: 10.1128/mbio.03545-23. Epub 2024 Feb 20.
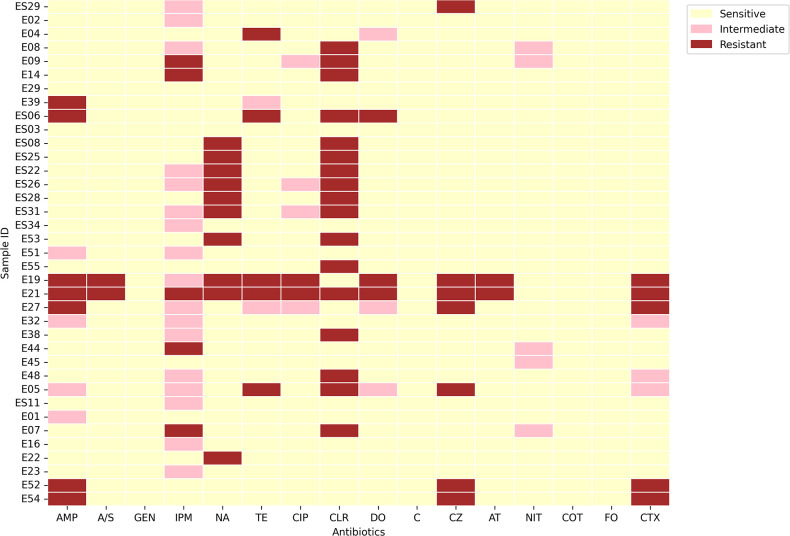
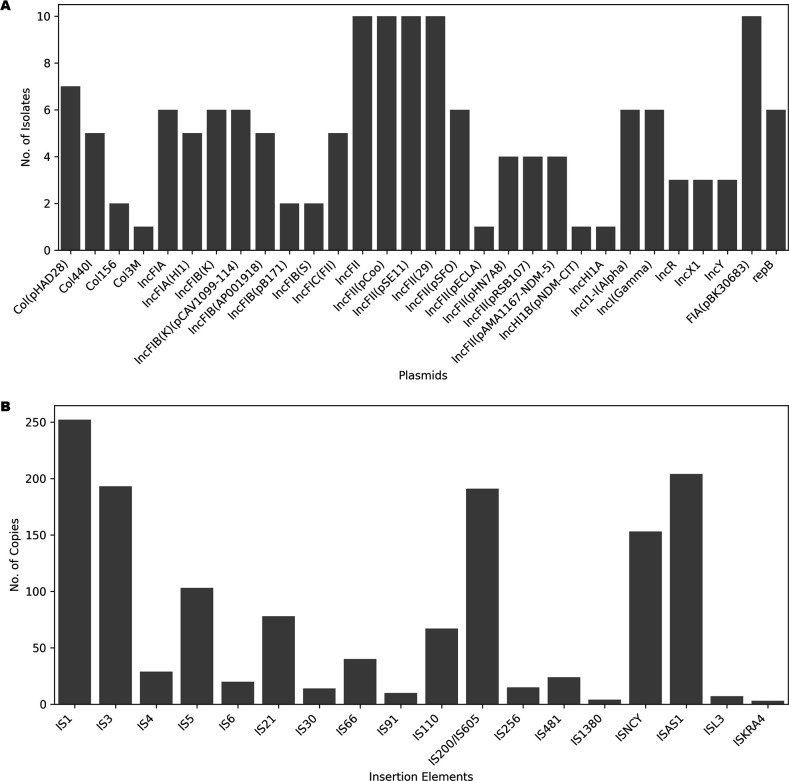
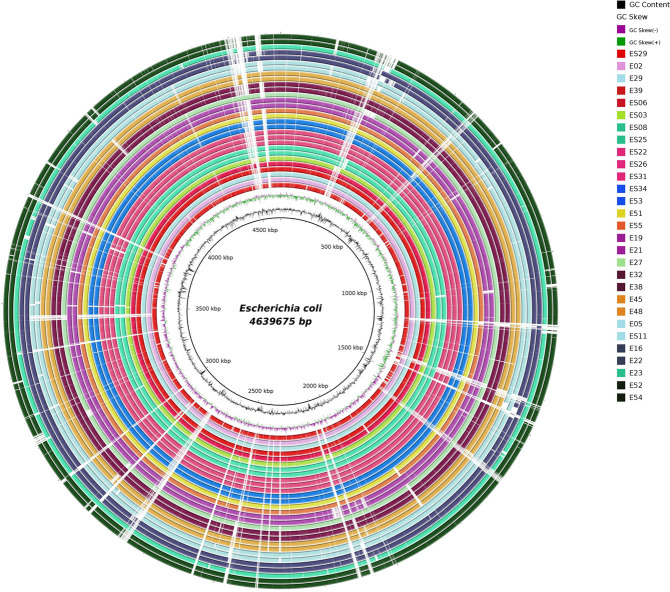
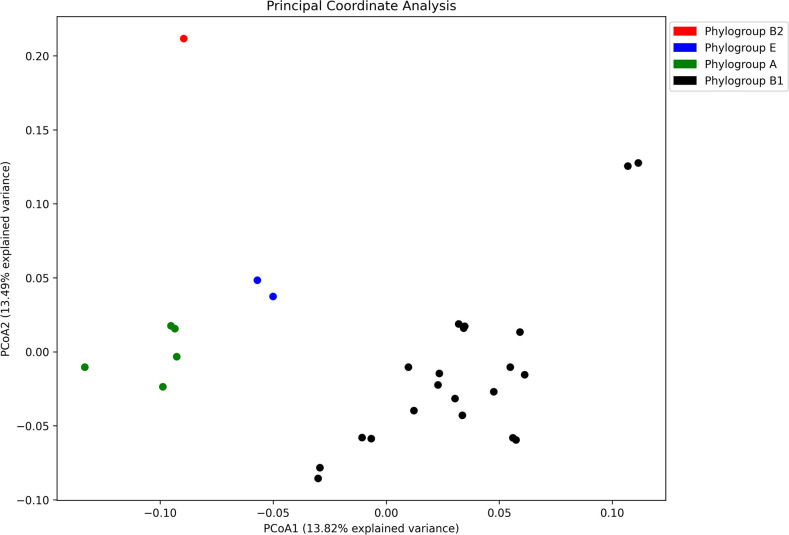
Populations of common commensal bacteria such as undergo genetic changes by the acquisition of certain virulence and antimicrobial resistance (AMR) encoding genetic elements leading to the emergence of pathogenic strains capable of surviving in the previously uninhabited or protected niches. These bacteria are also reported to be prevalent in the environment where they survive by adopting various recombination strategies to counter microflora of the soil and water, under constant selection pressure(s). In this study, we performed molecular characterization, phenotypic AMR analysis, and whole genome sequencing (WGS) of ( = 37) isolated from soil and surface water representing the urban and peri-urban areas. The primary aim of this study was to understand the genetic architecture and pathogenic acumen exhibited by environmental . WGS-based analysis entailing resistome and virulome profiling indicated the presence of various virulence (adherence, iron uptake, and toxins) and AMR encoding genes, including in the environmental isolates. A majority of our isolates belonged to phylogroup B1 (73%). A few isolates in our collection were of sequence type(s) (ST) 58 and 224 that could have emerged recently as clonal lineages and might pose risk of infection/transmission. Mobile genetic elements (MGEs) such as plasmids (predominantly) of the IncF family, prophages, pipolins, and insertion elements such as IS1 and IS5 were also observed to exist, which may presumably aid in the propagation of genes encoding resistance against antimicrobial drugs. The observed high prevalence of MGEs associated with multidrug resistance in pathogenic isolates belonging to the phylogroup B1 underscores the need for extended surveillance to keep track of and prevent the transmission of the bacterium to certain vulnerable human and animal populations.
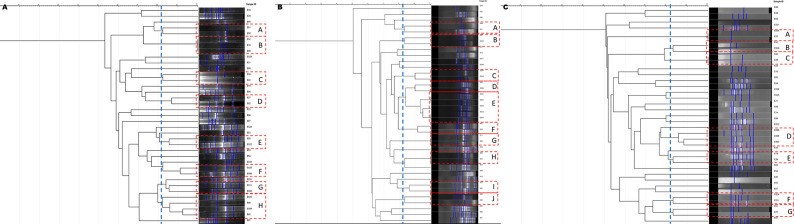
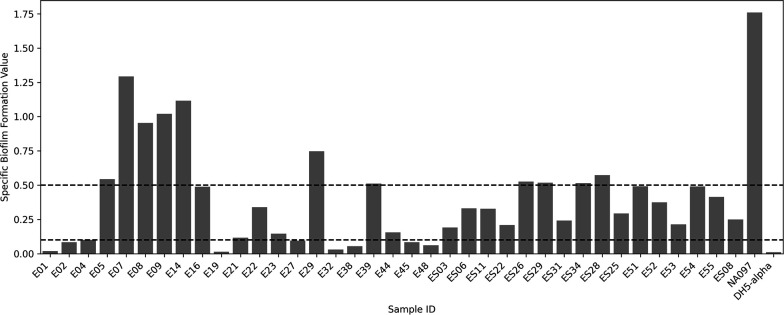
Evolutionary patterns of bacteria convey that they evolve into highly pathogenic forms by acquiring fitness advantages, such as AMR, and various virulence factors through the horizontal gene transfer (HGT)-mediated acquisition of MGEs. However, limited research on the genetic profiles of environmental , particularly from India, hinders our understanding of their transition to pathogenic forms and impedes the adoption of a comprehensive approach to address the connection between environmentally dwelling populations and human and veterinary public health. This study focuses on high-resolution genomic analysis of the environmental isolates aiming to understand the genetic similarities and differences among isolates from different environmental niches and uncover the survival strategies employed by these bacteria to thrive in their surroundings. Our approach involved molecular characterization of environmental samples using PCR-based DNA fingerprinting and subsequent WGS analysis. This multidisciplinary approach is likely to provide valuable insights into the understanding of any potential spill-over to human and animal populations and locales. Investigating these environmental isolates has significant potential for developing epidemiological strategies against transmission and understanding niche-specific evolutionary patterns.
常见共生细菌群体,如[具体细菌名称未给出],通过获取某些编码毒力和抗菌药物耐药性(AMR)的遗传元件而发生基因变化,导致能够在以前无人居住或受保护的生态位中存活的致病菌株出现。据报道,这些细菌在环境中也很普遍,在持续的选择压力下,它们通过采用各种重组策略来对抗土壤和水中的微生物群落从而生存。在本研究中,我们对从代表城市和城郊地区的土壤和地表水分离出的37株[具体细菌名称未给出]进行了分子特征分析、表型AMR分析和全基因组测序(WGS)。本研究的主要目的是了解环境中[具体细菌名称未给出]所展现的遗传结构和致病敏锐度。基于WGS的分析,包括耐药基因组和毒力基因组分析,表明在环境分离株中存在各种毒力(黏附、铁摄取和毒素)和AMR编码基因,包括[具体基因未给出]。我们的大多数分离株属于B1菌群(73%)。我们收集的一些分离株属于序列类型(ST)58和224,它们可能是最近作为克隆谱系出现的,可能会带来感染/传播风险。还观察到存在移动遗传元件(MGEs),如IncF家族的质粒(主要是)、前噬菌体、pipolins以及插入元件如IS1和IS5,这可能有助于编码抗抗菌药物抗性的基因传播。在属于B1菌群的致病性[具体细菌名称未给出]分离株中观察到与多药耐药性相关的MGEs的高流行率,这突出了需要进行扩展监测,以追踪并防止该细菌传播到某些易感染的人类和动物群体。
[具体细菌名称未给出]细菌的进化模式表明,它们通过水平基因转移(HGT)介导的MGEs获取,获得诸如AMR和各种毒力因子等适应性优势,从而进化为高致病性形式。然而,对环境中[具体细菌名称未给出]的遗传图谱的研究有限,特别是来自印度的研究,这阻碍了我们对它们向致病形式转变的理解,并阻碍了采用全面方法来解决环境中[具体细菌名称未给出]群体与人类和兽医公共卫生之间的联系。本研究专注于对环境中[具体细菌名称未给出]分离株进行高分辨率基因组分析,旨在了解来自不同环境生态位的分离株之间的遗传异同,并揭示这些细菌在其周围环境中生存所采用的策略。我们的方法包括使用基于PCR的DNA指纹图谱对环境样本进行分子特征分析以及随后的WGS分析。这种多学科方法可能会为理解任何潜在的向人类和动物群体及场所的溢出提供有价值的见解。研究这些环境分离株对于制定针对传播的流行病学策略和理解特定生态位进化模式具有重大潜力。