Institute for Molecular Bacteriology, TWINCORE Centre for Experimental and Clinical Infection Research, a joint venture between the Hannover Medical School (MHH) and the Helmholtz Centre for Infection Research (HZI), Hannover, Germany.
Cluster of Excellence RESIST (EXC 2155), Hannover Medical School (MHH), Hannover, Germany.
PLoS Genet. 2023 Aug 2;19(8):e1010842. doi: 10.1371/journal.pgen.1010842. eCollection 2023 Aug.
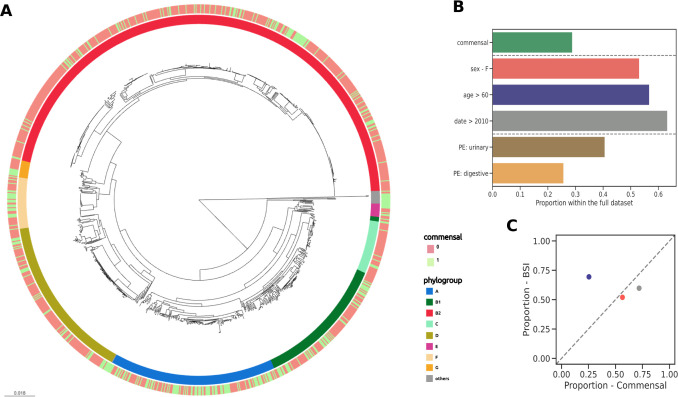
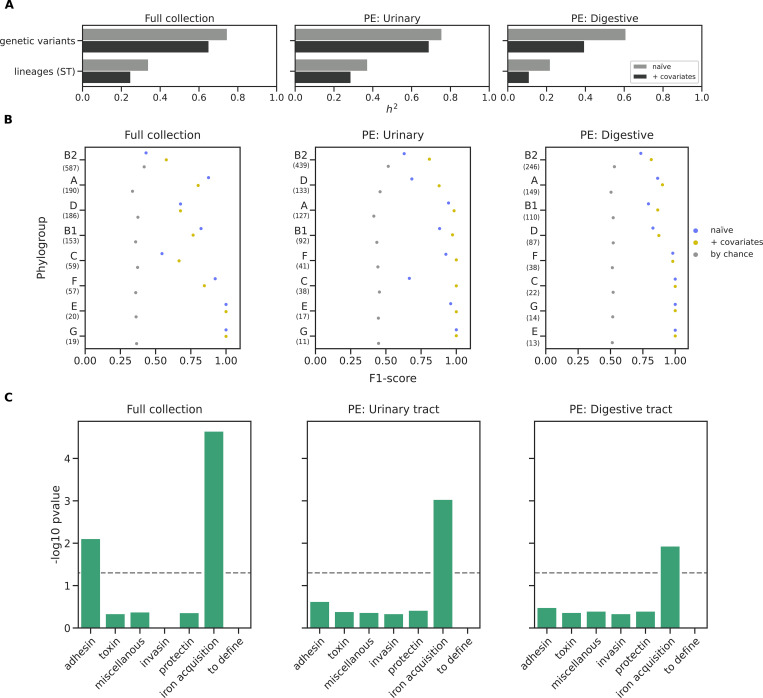
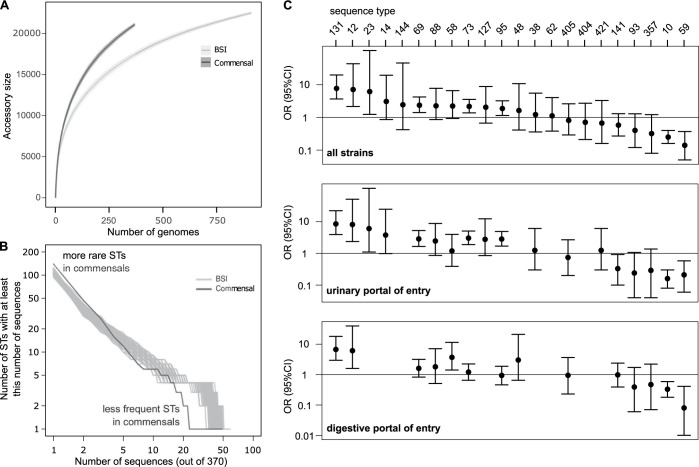
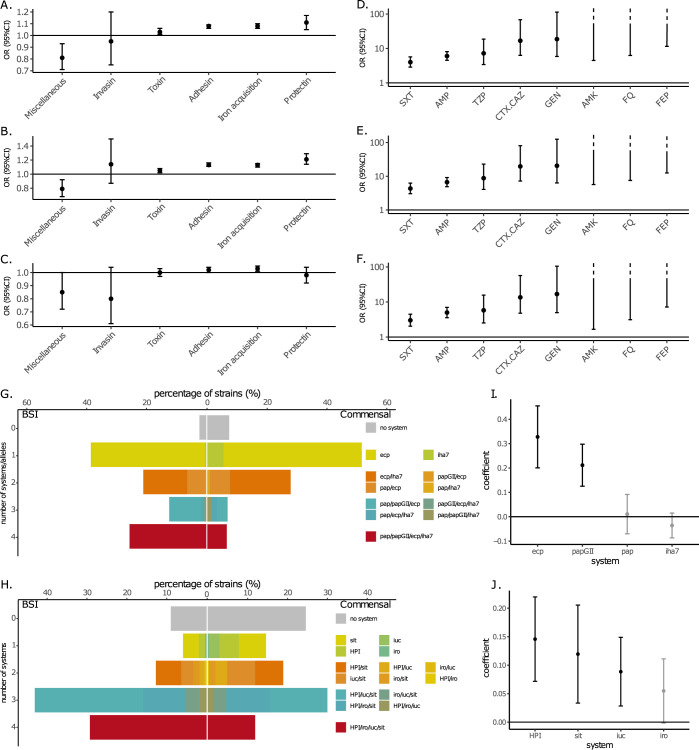
Escherichia coli is both a highly prevalent commensal and a major opportunistic pathogen causing bloodstream infections (BSI). A systematic analysis characterizing the genomic determinants of extra-intestinal pathogenic vs. commensal isolates in human populations, which could inform mechanisms of pathogenesis, diagnostic, prevention and treatment is still lacking. We used a collection of 912 BSI and 370 commensal E. coli isolates collected in France over a 17-year period (2000-2017). We compared their pangenomes, genetic backgrounds (phylogroups, STs, O groups), presence of virulence-associated genes (VAGs) and antimicrobial resistance genes, finding significant differences in all comparisons between commensal and BSI isolates. A machine learning linear model trained on all the genetic variants derived from the pangenome and controlling for population structure reveals similar differences in VAGs, discovers new variants associated with pathogenicity (capacity to cause BSI), and accurately classifies BSI vs. commensal strains. Pathogenicity is a highly heritable trait, with up to 69% of the variance explained by bacterial genetic variants. Lastly, complementing our commensal collection with an older collection from 1980, we predict that pathogenicity continuously increased through 1980, 2000, to 2010. Together our findings imply that E. coli exhibit substantial genetic variation contributing to the transition between commensalism and pathogenicity and that this species evolved towards higher pathogenicity.
大肠杆菌既是一种高度流行的共生菌,也是一种主要的机会致病菌,可引起血流感染(BSI)。目前仍缺乏系统分析,以描述人群中肠外致病性与共生分离株的基因组决定因素,这些因素可以为发病机制、诊断、预防和治疗提供信息。我们使用了 912 份血源分离的大肠杆菌和 370 份来自法国的共生大肠杆菌分离株的集合,这些分离株是在 17 年间(2000-2017 年)收集的。我们比较了它们的泛基因组、遗传背景( phylogroups、STs、O groups)、毒力相关基因(VAGs)和抗生素耐药基因,发现共生和血源分离株之间的所有比较都存在显著差异。基于泛基因组和群体结构控制,对所有遗传变异进行训练的机器学习线性模型揭示了 VAGs 之间的相似差异,发现了与致病性(引起 BSI 的能力)相关的新变异,并准确地对 BSI 与共生菌株进行分类。致病性是一种高度可遗传的特征,高达 69%的变异可以由细菌遗传变异来解释。最后,我们用 1980 年的一个更老的共生分离株集合来补充我们的共生分离株集合,我们预测致病性在 1980 年、2000 年和 2010 年之间持续增加。总的来说,我们的研究结果表明,大肠杆菌表现出大量的遗传变异,有助于从共生到致病性的转变,并且该物种朝着更高的致病性进化。