Singh Ritambhara, Demetci Pinar, Bonora Giancarlo, Ramani Vijay, Lee Choli, Fang He, Duan Zhijun, Deng Xinxian, Shendure Jay, Disteche Christine, Noble William Stafford
Department of Computer Science, Center for Computational Molecular Biology, Brown University.
Center for Computational Molecular Biology, Brown University.
ACM BCB. 2020 Sep;2020:1-10. doi: 10.1145/3388440.3412410.
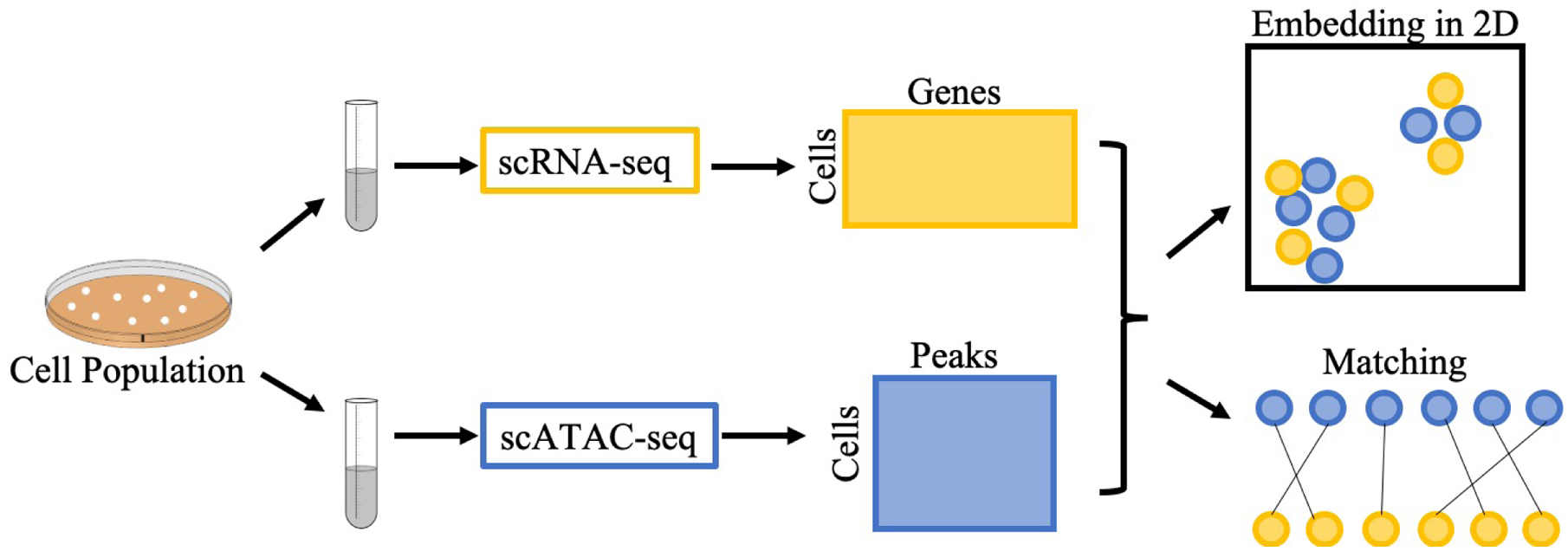
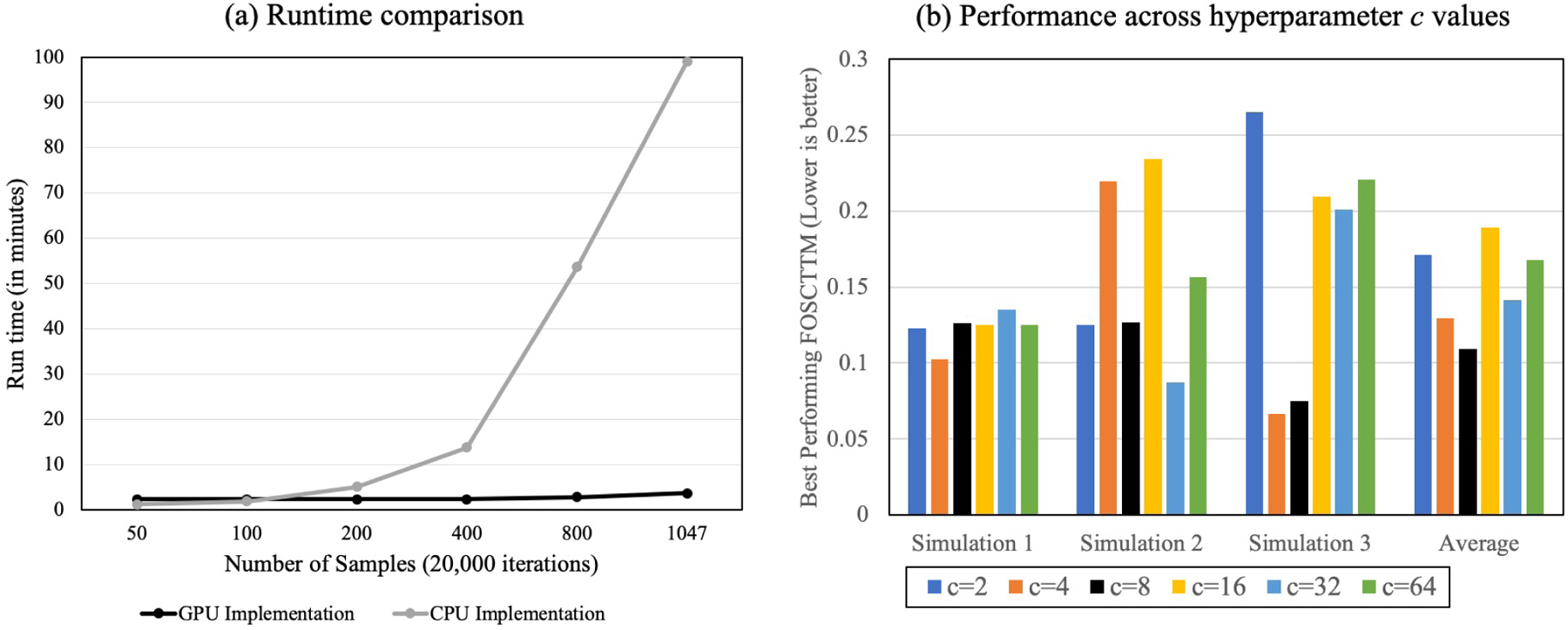
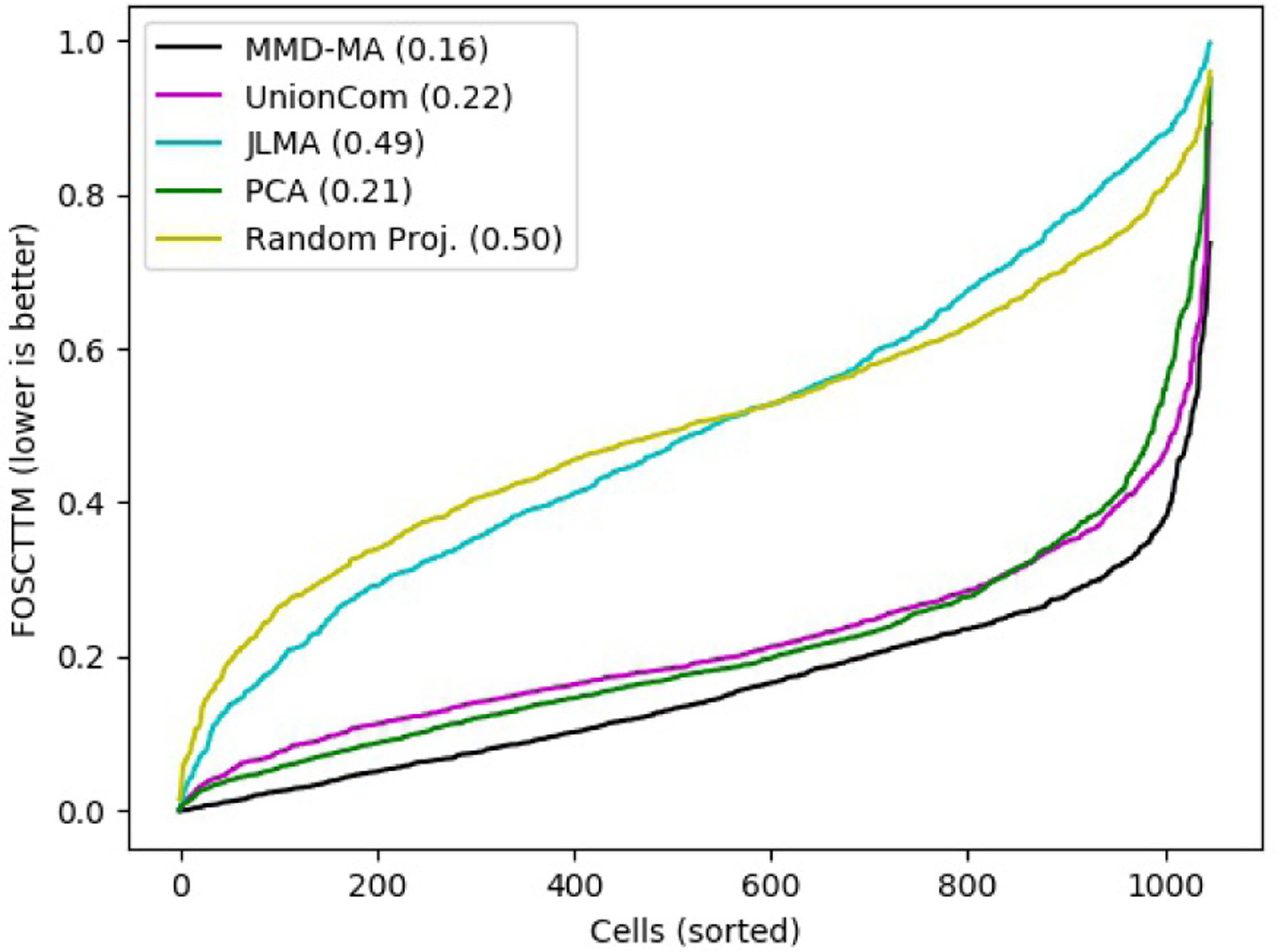
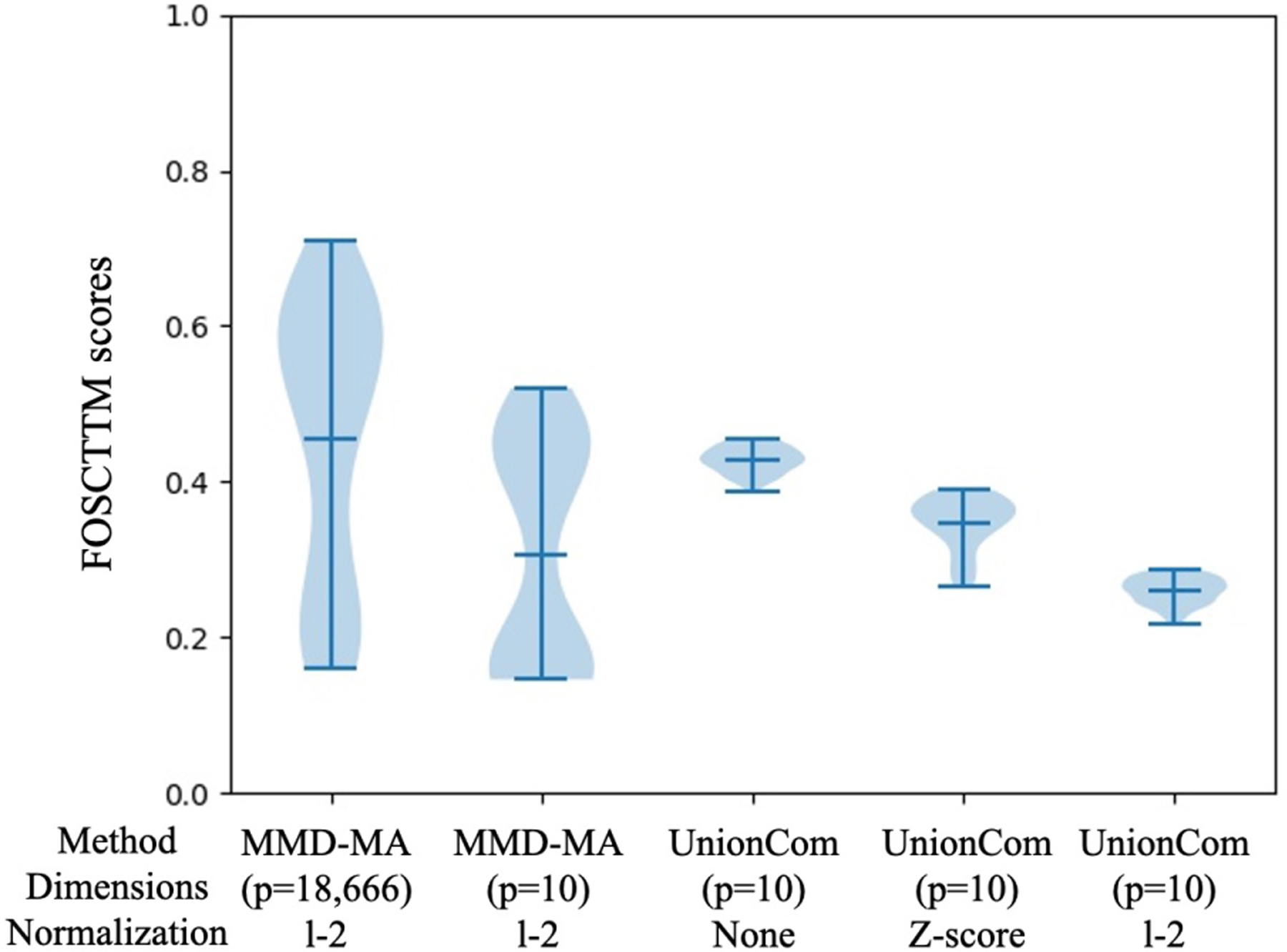
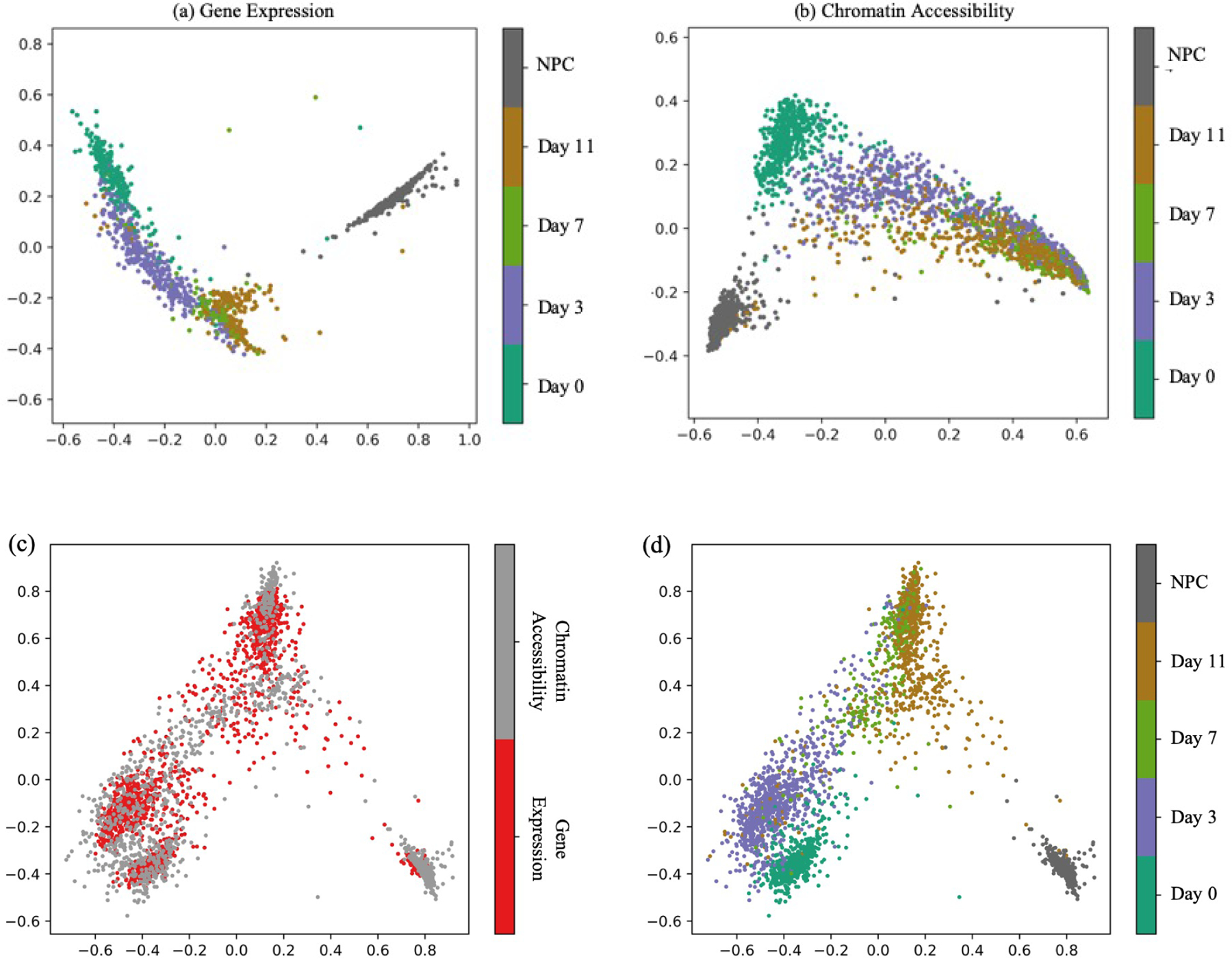
Integrating single-cell measurements that capture different properties of the genome is vital to extending our understanding of genome biology. This task is challenging due to the lack of a shared axis across datasets obtained from different types of single-cell experiments. For most such datasets, we lack corresponding information among the cells (samples) and the measurements (features). In this scenario, unsupervised algorithms that are capable of aligning single-cell experiments are critical to learning an co-assay that can help draw correspondences among the cells. Maximum mean discrepancy-based manifold alignment (MMD-MA) is such an unsupervised algorithm. Without requiring correspondence information, it can align single-cell datasets from different modalities in a common shared latent space, showing promising results on simulations and a small-scale single-cell experiment with 61 cells. However, it is essential to explore the applicability of this method to larger single-cell experiments with thousands of cells so that it can be of practical interest to the community. In this paper, we apply MMD-MA to two recent datasets that measure transcriptome and chromatin accessibility in ~2000 single cells. To scale the runtime of MMD-MA to a more substantial number of cells, we extend the original implementation to run on GPUs. We also introduce a method to automatically select one of the user-defined parameters, thus reducing the hyperparameter search space. We demonstrate that the proposed extensions allow MMD-MA to accurately align state-of-the-art single-cell experiments.
整合能够捕捉基因组不同特性的单细胞测量数据对于拓展我们对基因组生物学的理解至关重要。由于缺乏从不同类型的单细胞实验获得的数据集之间的共享轴,这项任务具有挑战性。对于大多数此类数据集,我们在细胞(样本)和测量(特征)之间缺乏相应信息。在这种情况下,能够对齐单细胞实验的无监督算法对于学习一种有助于在细胞之间建立对应关系的联合分析至关重要。基于最大均值差异的流形对齐(MMD-MA)就是这样一种无监督算法。它无需对应信息,就能在一个共同的共享潜在空间中对齐来自不同模态的单细胞数据集,在模拟和一个包含61个细胞的小规模单细胞实验中显示出了有前景的结果。然而,探索这种方法在包含数千个细胞的更大规模单细胞实验中的适用性至关重要,这样它才能引起该领域的实际兴趣。在本文中,我们将MMD-MA应用于两个最近的数据集,这些数据集测量了约2000个单细胞中的转录组和染色质可及性。为了将MMD-MA的运行时间扩展到更多细胞,我们将原始实现扩展为在GPU上运行。我们还引入了一种方法来自动选择用户定义的参数之一,从而减少超参数搜索空间。我们证明,所提出的扩展允许MMD-MA准确对齐最新的单细胞实验。