Guo Zhiye, Wu Tianqi, Liu Jian, Hou Jie, Cheng Jianlin
Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, MO 65211, USA.
Department of Computer Science, Saint Louis University, St. Louis, MO 63103, USA.
Bioinformatics. 2021 Oct 11;37(19):3190-3196. doi: 10.1093/bioinformatics/btab355.
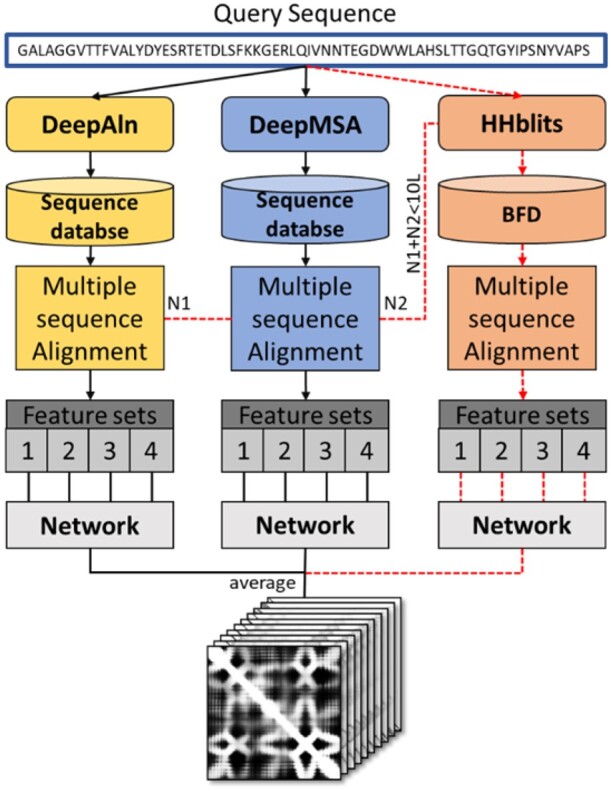
Accurate prediction of residue-residue distances is important for protein structure prediction. We developed several protein distance predictors based on a deep learning distance prediction method and blindly tested them in the 14th Critical Assessment of Protein Structure Prediction (CASP14). The prediction method uses deep residual neural networks with the channel-wise attention mechanism to classify the distance between every two residues into multiple distance intervals. The input features for the deep learning method include co-evolutionary features as well as other sequence-based features derived from multiple sequence alignments (MSAs). Three alignment methods are used with multiple protein sequence/profile databases to generate MSAs for input feature generation. Based on different configurations and training strategies of the deep learning method, five MULTICOM distance predictors were created to participate in the CASP14 experiment.
Benchmarked on 37 hard CASP14 domains, the best performing MULTICOM predictor is ranked 5th out of 30 automated CASP14 distance prediction servers in terms of precision of top L/5 long-range contact predictions [i.e. classifying distances between two residues into two categories: in contact (<8 Angstrom) and not in contact otherwise] and performs better than the best CASP13 distance prediction method. The best performing MULTICOM predictor is also ranked 6th among automated server predictors in classifying inter-residue distances into 10 distance intervals defined by CASP14 according to the precision of distance classification. The results show that the quality and depth of MSAs depend on alignment methods and sequence databases and have a significant impact on the accuracy of distance prediction. Using larger training datasets and multiple complementary features improves prediction accuracy. However, the number of effective sequences in MSAs is only a weak indicator of the quality of MSAs and the accuracy of predicted distance maps. In contrast, there is a strong correlation between the accuracy of contact/distance predictions and the average probability of the predicted contacts, which can therefore be more effectively used to estimate the confidence of distance predictions and select predicted distance maps.
The software package, source code and data of DeepDist2 are freely available at https://github.com/multicom-toolbox/deepdist and https://zenodo.org/record/4712084#.YIIM13VKhQM.
Supplementary data are available at Bioinformatics online.
准确预测残基间距离对于蛋白质结构预测至关重要。我们基于深度学习距离预测方法开发了几种蛋白质距离预测器,并在第14届蛋白质结构预测关键评估(CASP14)中对它们进行了盲测。该预测方法使用带有通道注意力机制的深度残差神经网络,将每两个残基之间的距离分类到多个距离区间。深度学习方法的输入特征包括共进化特征以及从多序列比对(MSA)中衍生的其他基于序列的特征。使用三种比对方法与多个蛋白质序列/谱数据库来生成用于输入特征生成的MSA。基于深度学习方法的不同配置和训练策略,创建了五个MULTICOM距离预测器来参与CASP14实验。
以37个CASP14硬结构域为基准,表现最佳的MULTICOM预测器在顶级L/5远程接触预测的精度方面(即将两个残基之间的距离分为两类:接触(<8埃)和不接触),在30个自动化CASP14距离预测服务器中排名第5,并且比最佳的CASP13距离预测方法表现更好。在根据距离分类精度将残基间距离分类为CASP14定义的10个距离区间方面,表现最佳的MULTICOM预测器在自动化服务器预测器中也排名第6。结果表明,MSA的质量和深度取决于比对方法和序列数据库,并且对距离预测的准确性有重大影响。使用更大的训练数据集和多个互补特征可提高预测准确性。然而,MSA中有效序列的数量只是MSA质量和预测距离图准确性的一个弱指标。相比之下,接触/距离预测的准确性与预测接触的平均概率之间存在很强的相关性,因此可以更有效地用于估计距离预测的置信度并选择预测距离图。
DeepDist2的软件包、源代码和数据可在https://github.com/multicom-toolbox/deepdist和https://zenodo.org/record/4712084#.YIIM13VKhQM上免费获取。
补充数据可在《生物信息学》在线获取。