Ascari Giulia, Rendtorff Nanna D, De Bruyne Marieke, De Zaeytijd Julie, Van Lint Michel, Bauwens Miriam, Van Heetvelde Mattias, Arno Gavin, Jacob Julie, Creytens David, Van Dorpe Jo, Van Laethem Thalia, Rosseel Toon, De Pooter Tim, De Rijk Peter, De Coster Wouter, Menten Björn, Rey Alfredo Dueñas, Strazisar Mojca, Bertelsen Mette, Tranebjaerg Lisbeth, De Baere Elfride
Center for Medical Genetics Ghent, Ghent University Hospital, Ghent, Belgium.
Department of Biomolecular Medicine, Ghent University, Ghent, Belgium.
Front Cell Dev Biol. 2021 Apr 21;9:664317. doi: 10.3389/fcell.2021.664317. eCollection 2021.
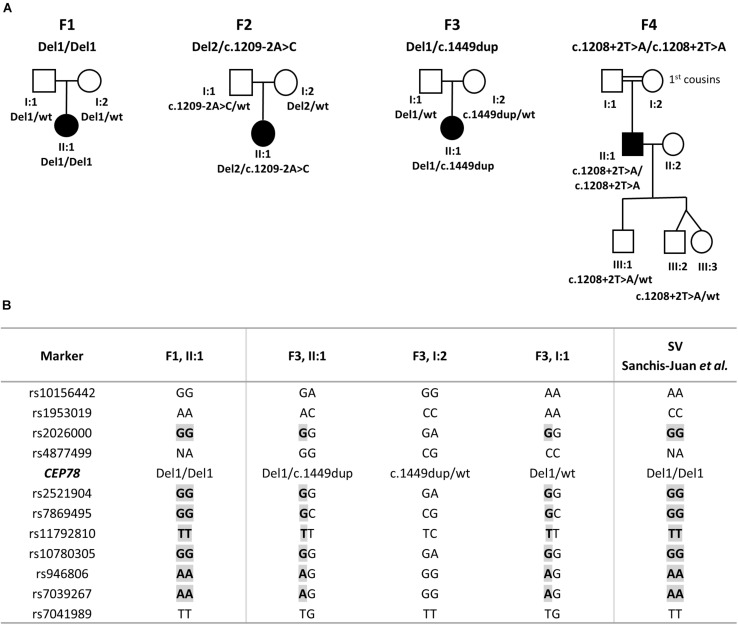
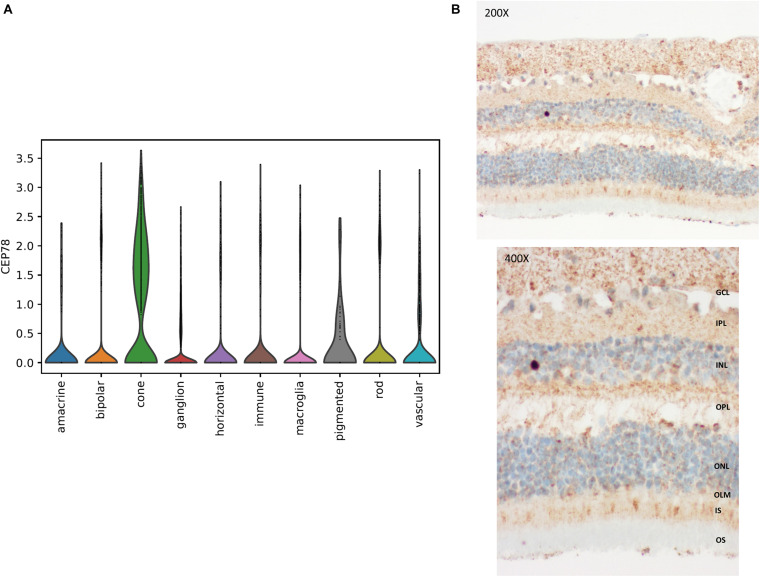
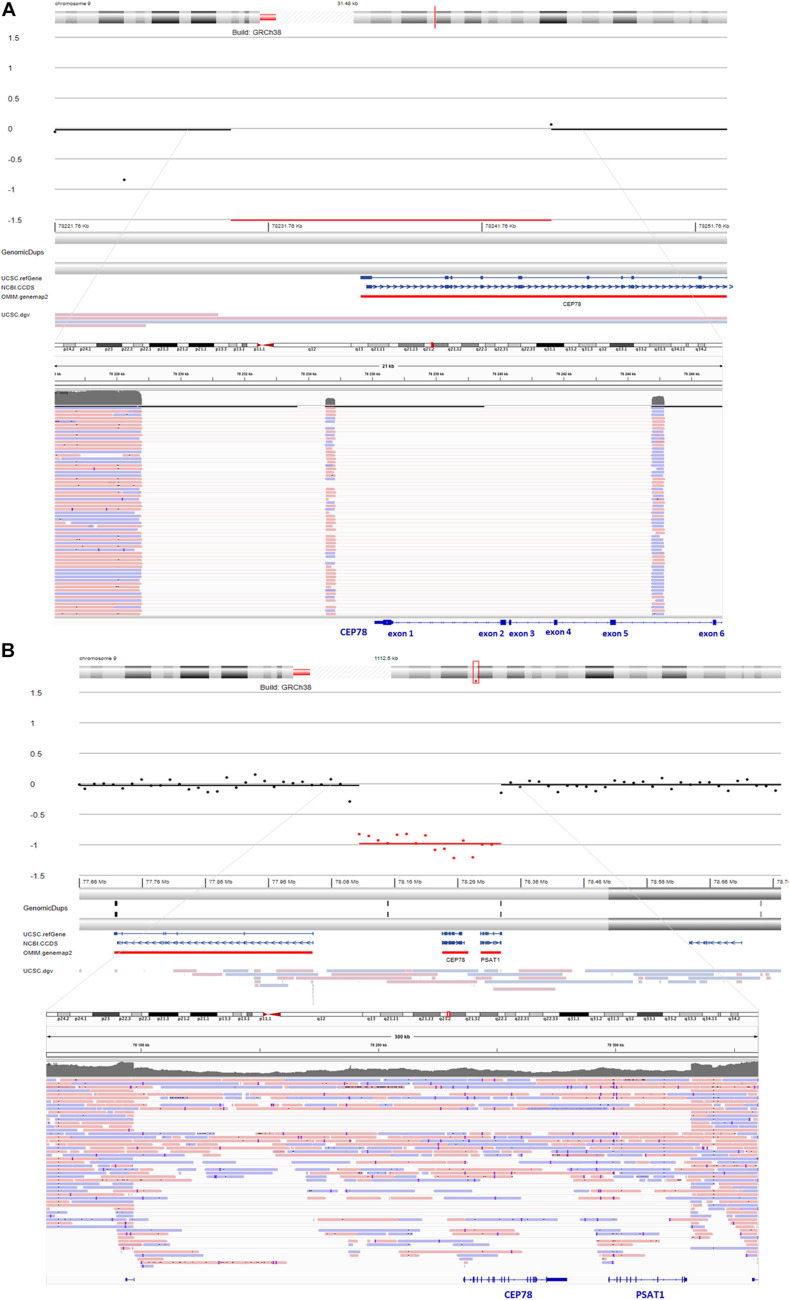
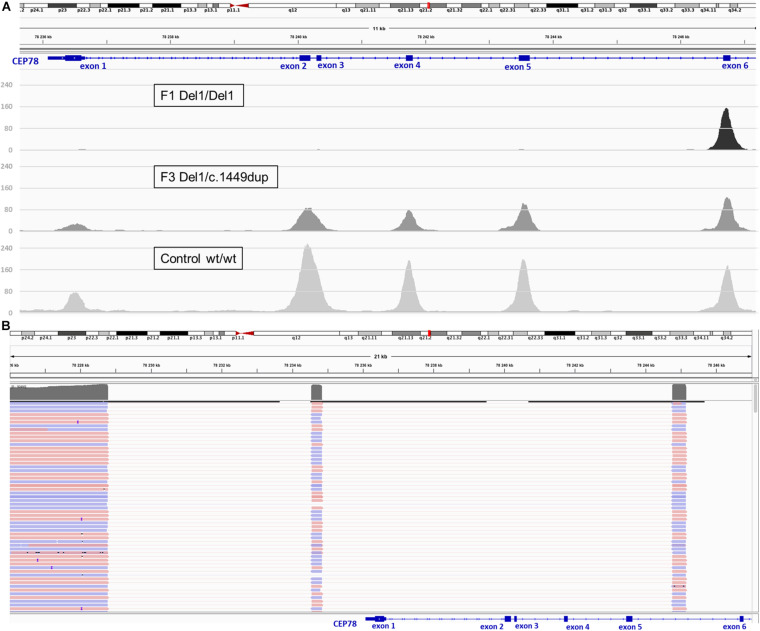
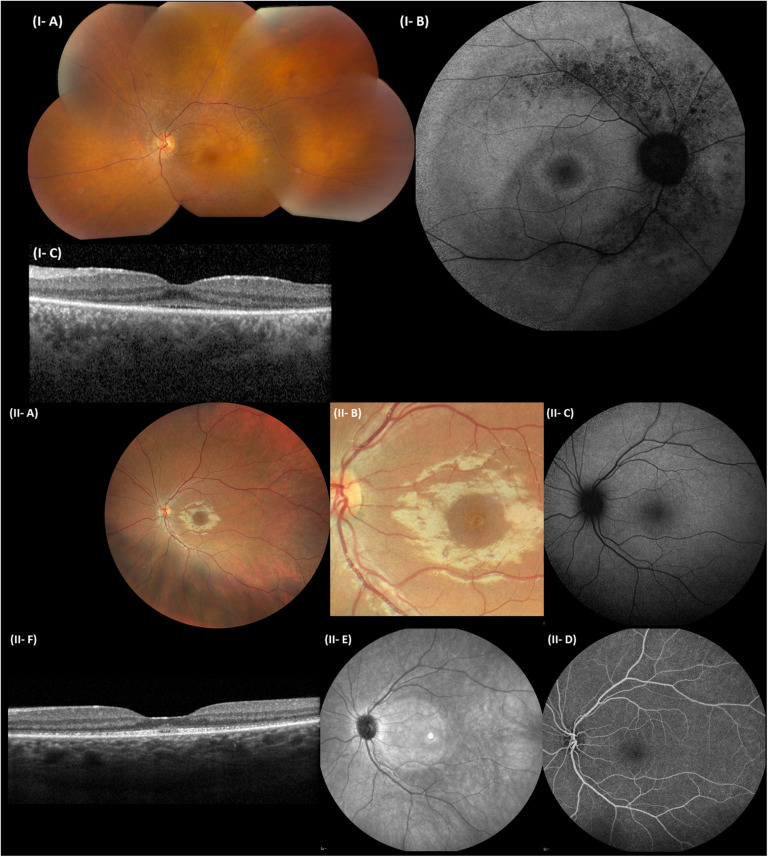
Inactivating variants as well as a missense variant in the centrosomal gene have been identified in autosomal recessive cone-rod dystrophy with hearing loss (CRDHL), a rare syndromic inherited retinal disease distinct from Usher syndrome. Apart from this, a complex structural variant (SV) implicating has been reported in CRDHL. Here we aimed to expand the genetic architecture of typical CRDHL by the identification of complex SVs of the region and characterization of their underlying mechanisms. Approaches used for the identification of the SVs are shallow whole-genome sequencing (sWGS) combined with quantitative polymerase chain reaction (PCR) and long-range PCR, or ExomeDepth analysis on whole-exome sequencing (WES) data. Targeted or whole-genome nanopore long-read sequencing (LRS) was used to delineate breakpoint junctions at the nucleotide level. For all SVs cases, the effect of the SVs on expression was assessed using quantitative PCR on patient-derived RNA. Apart from two novel canonical splice variants and a frameshifting single-nucleotide variant (SNV), two SVs affecting were identified in three unrelated individuals with CRDHL: a heterozygous total gene deletion of 235 kb and a partial gene deletion of 15 kb in a heterozygous and homozygous state, respectively. Assessment of the molecular consequences of the SVs on patient's materials displayed a loss-of-function effect. Delineation and characterization of the 15-kb deletion using targeted LRS revealed the previously described complex SV, suggestive of a recurrent genomic rearrangement. A founder haplotype was demonstrated for the latter SV in cases of Belgian and British origin, respectively. The novel 235-kb deletion was delineated using whole-genome LRS. Breakpoint analysis showed microhomology and pointed to a replication-based underlying mechanism. Moreover, data mining of bulk and single-cell human and mouse transcriptional datasets, together with CEP78 immunostaining on human retina, linked the CEP78 expression domain with its phenotypic manifestations. Overall, this study supports that the locus is prone to distinct SVs and that SV analysis should be considered in a genetic workup of CRDHL. Finally, it demonstrated the power of sWGS and both targeted and whole-genome LRS in identifying and characterizing complex SVs in patients with ocular diseases.
在常染色体隐性遗传性视锥-视杆营养不良伴听力丧失(CRDHL)中已鉴定出中心体基因的失活变异以及一个错义变异,CRDHL是一种罕见的综合征性遗传性视网膜疾病,与Usher综合征不同。除此之外,在CRDHL中还报道了一个涉及 的复杂结构变异(SV)。在这里,我们旨在通过鉴定该区域的复杂SV并表征其潜在机制,来扩展典型CRDHL的遗传结构。用于鉴定SV的方法包括浅层全基因组测序(sWGS)结合定量聚合酶链反应(PCR)和长程PCR,或对全外显子组测序(WES)数据进行ExomeDepth分析。使用靶向或全基因组纳米孔长读长测序(LRS)在核苷酸水平上描绘断点连接。对于所有SV病例,使用患者来源的RNA进行定量PCR来评估SV对 表达的影响。除了两个新的典型 剪接变异和一个移码单核苷酸变异(SNV)外,在三名无关的CRDHL患者中鉴定出两个影响 的SV:一个235 kb的杂合性全基因缺失和一个分别处于杂合和纯合状态的15 kb部分基因缺失。对患者材料中SV的分子后果评估显示出功能丧失效应。使用靶向LRS对15 kb缺失进行描绘和表征,揭示了先前描述的复杂 SV,提示存在反复发生的基因组重排。分别在比利时和英国血统的病例中证明了后一种SV的奠基者单倍型。使用全基因组LRS描绘了新的235 kb缺失。断点分析显示出微同源性,并指向基于复制的潜在机制。此外,对大量和单细胞人类及小鼠转录数据集的数据挖掘,以及对人类视网膜进行的CEP78免疫染色,将CEP78表达域与其表型表现联系起来。总体而言,本研究支持 基因座易于出现不同的SV,并且在CRDHL的基因检查中应考虑SV分析。最后,它展示了sWGS以及靶向和全基因组LRS在鉴定和表征眼部疾病患者复杂SV方面的能力。