Trinh Hong Son, Luong Tuan Hiep, Lai Thanh Tung, Nguyen Thanh Khiem
Department of Oncology, Viet Duc University Hospital, Hanoi, Viet Nam.
Department of Surgery, Hanoi Medical University, Hanoi, Viet Nam.
Int J Surg Case Rep. 2021 Jun;83:105951. doi: 10.1016/j.ijscr.2021.105951. Epub 2021 Apr 30.
Hepatoid carcinoma (HC) is a rare type of malignant tumor that shared similar features of morphology and immunohistochemistry with hepatocellular carcinoma (HCC). Pancreatic HC exists as either pure or mixed type. Mixed pancreatic HC is extremely rare, with only a few cases reported in the literature to date. Because of the rarity of mixed pancreatic HC, its clinical features including incidence, characteristics, and prognosis remain unclear. We herein report a case of a 49-year-old man who was diagnosed with mixed pancreatic HC with neuroendocrine differentiation and was treated with pancreaticoduodenectomy and adjuvant chemotherapy. We also review the existing case reports in literature.
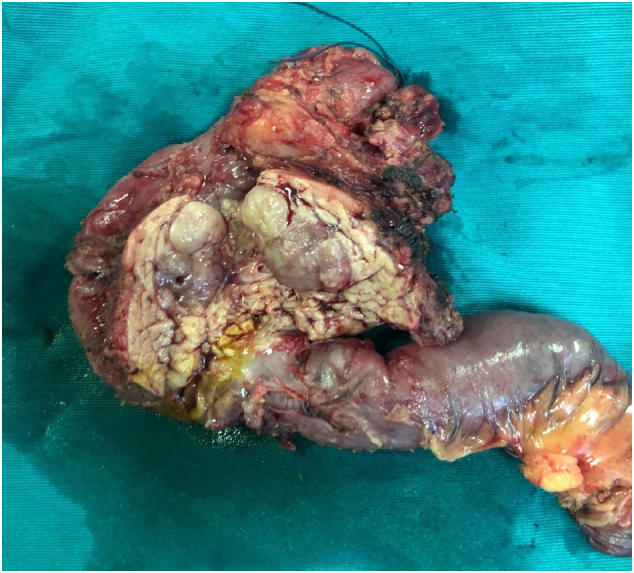
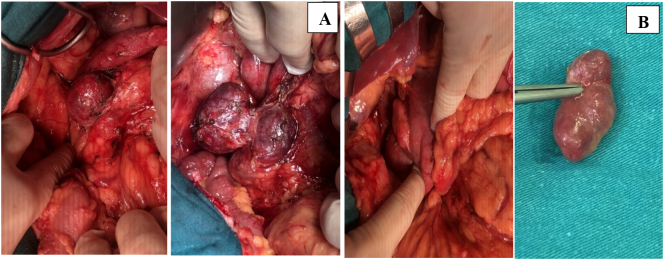
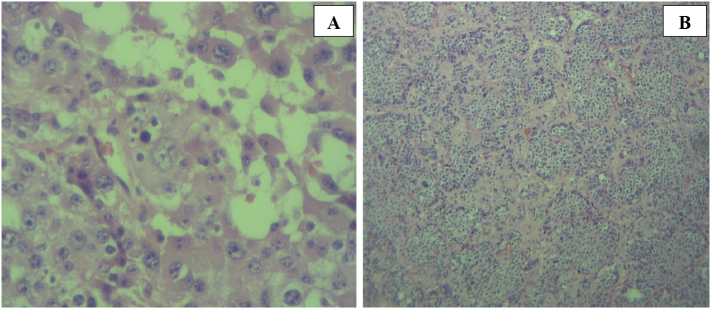
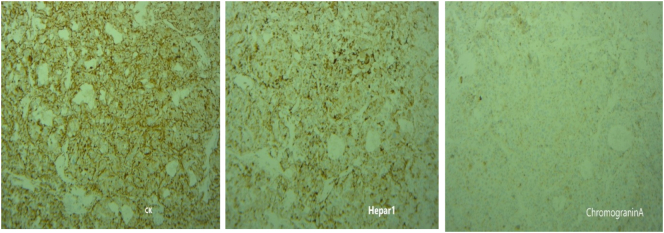
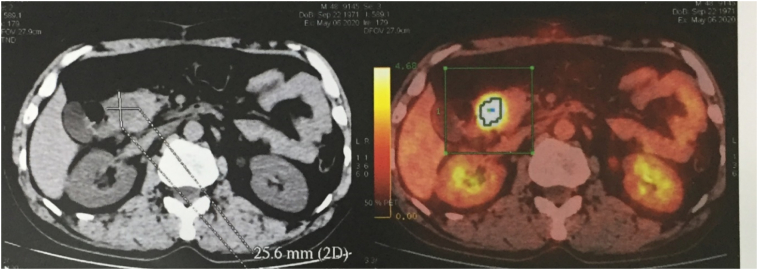
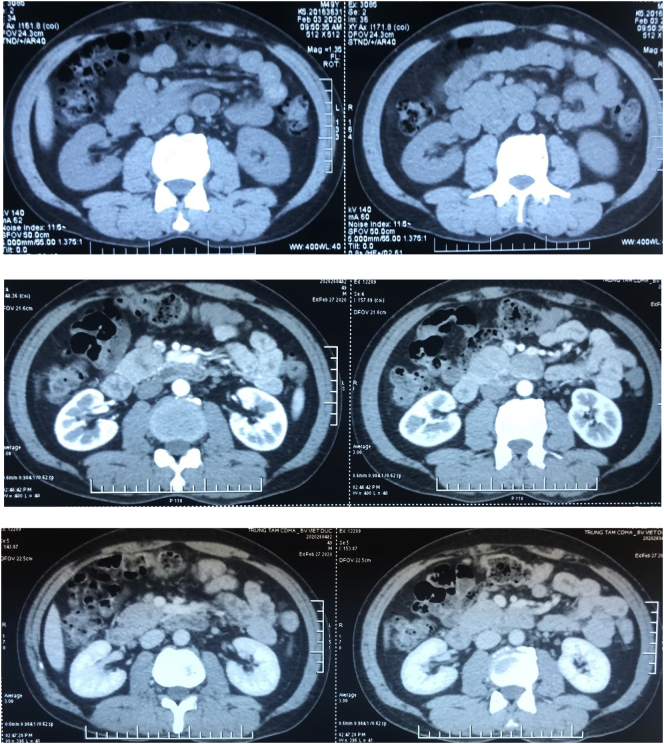
A 49-year-old man was admitted to our hospital after a chronic abdominal pain in the upper right quadrant. Abdominal ultrasound revealed only one low-density retroperitoneal mass measured at 20 × 48mm in size in the pancreatic-duodenal junction, whereas contrast-enhanced computed tomography (CT) revealed three lymphatic neoplasms measured at 28 × 22 × 30 mm, 27 × 33 × 38 mm and 22 × 35 × 48 mm in size in the retroperitoneal pancreatic-duodenal junction. Ultrasound-guided tumor biopsy was performed. Pathological reading of tumor biopsy suspected of Paraganglioma/pheochromocytoma. Laparotomic retroperitoneal tumoral resection and lymphadenectomy was then performed. Histological reading was lymphatic metastasis of primary pancreatic hepatocellular carcinoma with neuroendocrine differentiation, which were immunohistochemically positive for CKAE1/AE3, Hepatocyte paraffin 1, Chromogranin. After three weeks of the first surgery, the patient was assigned with Positron Emission Tomography - Computed Tomography (PET-CT) before adjuvant chemotherapy, revealing a low-density high-metabolism mass, 26 × 28 mm in size within the parenchyma of pancreatic head. Laparotomic pancreaticoduodenectomy and standard lymphadenectomy was performed to resect one mass, which revealed the same immunohistology features with the first mass. The patient was followed up with FOLFIRINOX protocol, and after 12 cycles, there was no evidence of postoperative recurrence.
There are few reported cases describing pancreatic hepatoid carcinoma, especially mixed form with other histological associated component. Neuroendocrine differentiation is the majority associated component with 62.5% of all cases of mixed - type form.
Primary pancreatic hepatocellular carcinoma with neuroendocrine differentiation was rare, biopsy and immunohistochemistry appeared with high diagnostic value in this case. The prognosis of pancreatic HC depends on the extent and tumor eradication, and in this case we recorded no postoperative complications and no recurrence in the 6-month follow-up period.
肝样腺癌(HC)是一种罕见的恶性肿瘤,在形态学和免疫组化方面与肝细胞癌(HCC)具有相似特征。胰腺HC可分为纯型或混合型。混合性胰腺HC极为罕见,迄今为止文献中仅报道了少数病例。由于混合性胰腺HC的罕见性,其临床特征,包括发病率、特点和预后仍不明确。我们在此报告一例49岁男性,被诊断为具有神经内分泌分化的混合性胰腺HC,并接受了胰十二指肠切除术和辅助化疗。我们还回顾了文献中现有的病例报告。
一名49岁男性因右上腹慢性疼痛入院。腹部超声仅显示在胰十二指肠交界处有一个大小为20×48mm的低密度腹膜后肿块,而增强计算机断层扫描(CT)显示在腹膜后胰十二指肠交界处有三个大小分别为28×22×30mm、27×33×38mm和22×35×48mm的淋巴瘤。进行了超声引导下肿瘤活检。肿瘤活检的病理检查怀疑为副神经节瘤/嗜铬细胞瘤。随后进行了剖腹腹膜后肿瘤切除术和淋巴结清扫术。组织学检查为原发性胰腺肝细胞癌伴神经内分泌分化的淋巴结转移,免疫组化检查CKAE1/AE3、肝细胞石蜡1、嗜铬粒蛋白呈阳性。首次手术后三周,患者在辅助化疗前接受了正电子发射断层扫描 - 计算机断层扫描(PET-CT)检查,显示胰头实质内有一个大小为26×28mm的低密度高代谢肿块。进行了剖腹胰十二指肠切除术和标准淋巴结清扫术以切除一个肿块,其免疫组织学特征与第一个肿块相同。患者采用FOLFIRINOX方案进行随访,12个周期后,无术后复发迹象。
报道的胰腺肝样腺癌病例很少,尤其是与其他组织学相关成分的混合形式。神经内分泌分化是混合类型所有病例中最主要的相关成分,占62.5%。
原发性胰腺肝细胞癌伴神经内分泌分化罕见,活检和免疫组化在该病例中具有较高的诊断价值。胰腺HC的预后取决于病变范围和肿瘤清除情况,在本病例中,我们在6个月的随访期内未记录到术后并发症和复发情况。