Department of Pediatrics, Columbia University Irving Medical Center, 1150 St. Nicholas Avenue, Room 620, New York, NY, 10032, USA.
Department of Systems Biology, Columbia University, New York, NY, USA.
Genome Med. 2021 May 10;13(1):80. doi: 10.1186/s13073-021-00891-1.
Pulmonary arterial hypertension (PAH) is a lethal vasculopathy characterized by pathogenic remodeling of pulmonary arterioles leading to increased pulmonary pressures, right ventricular hypertrophy, and heart failure. PAH can be associated with other diseases (APAH: connective tissue diseases, congenital heart disease, and others) but often the etiology is idiopathic (IPAH). Mutations in bone morphogenetic protein receptor 2 (BMPR2) are the cause of most heritable cases but the vast majority of other cases are genetically undefined.
To identify new risk genes, we utilized an international consortium of 4241 PAH cases with exome or genome sequencing data from the National Biological Sample and Data Repository for PAH, Columbia University Irving Medical Center, and the UK NIHR BioResource - Rare Diseases Study. The strength of this combined cohort is a doubling of the number of IPAH cases compared to either national cohort alone. We identified protein-coding variants and performed rare variant association analyses in unrelated participants of European ancestry, including 1647 IPAH cases and 18,819 controls. We also analyzed de novo variants in 124 pediatric trios enriched for IPAH and APAH-CHD.
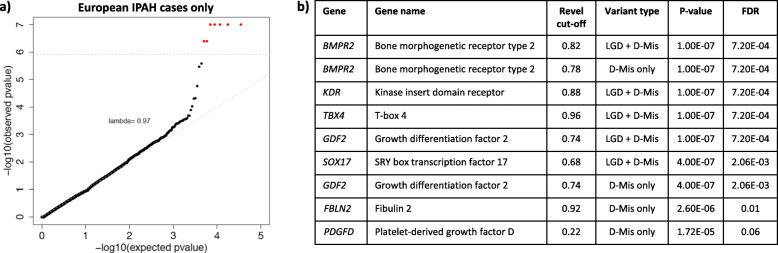
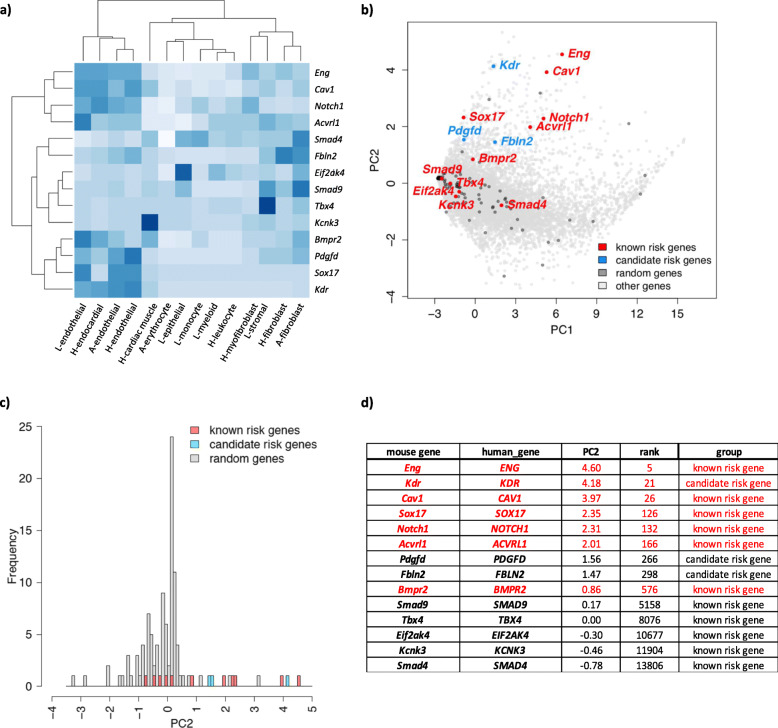
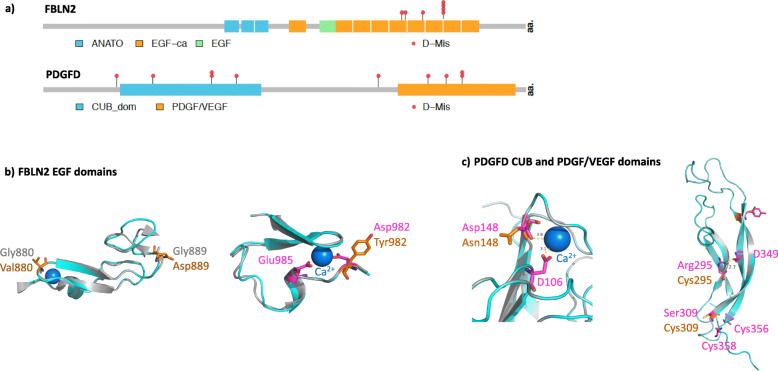
Seven genes with rare deleterious variants were associated with IPAH with false discovery rate smaller than 0.1: three known genes (BMPR2, GDF2, and TBX4), two recently identified candidate genes (SOX17, KDR), and two new candidate genes (fibulin 2, FBLN2; platelet-derived growth factor D, PDGFD). The new genes were identified based solely on rare deleterious missense variants, a variant type that could not be adequately assessed in either cohort alone. The candidate genes exhibit expression patterns in lung and heart similar to that of known PAH risk genes, and most variants occur in conserved protein domains. For pediatric PAH, predicted deleterious de novo variants exhibited a significant burden compared to the background mutation rate (2.45×, p = 2.5e-5). At least eight novel pediatric candidate genes carrying de novo variants have plausible roles in lung/heart development.
Rare variant analysis of a large international consortium identified two new candidate genes-FBLN2 and PDGFD. The new genes have known functions in vasculogenesis and remodeling. Trio analysis predicted that ~ 15% of pediatric IPAH may be explained by de novo variants.
肺动脉高压(PAH)是一种致命的血管病变,其特征为肺小动脉发生病理性重构,导致肺压升高、右心室肥厚和心力衰竭。PAH 可与其他疾病相关(APAH:结缔组织疾病、先天性心脏病等),但病因通常是特发性的(IPAH)。骨形态发生蛋白受体 2(BMPR2)的突变是大多数遗传性病例的原因,但绝大多数其他病例的遗传原因尚未确定。
为了确定新的风险基因,我们利用了一个国际联盟的 4241 例 PAH 病例,这些病例来自国家生物样本和数据资源库的 PAH、哥伦比亚大学欧文医学中心和英国国家卫生研究院生物资源——罕见疾病研究的外显子或基因组测序数据。与任何一个国家队列相比,该联合队列的 IPAH 病例数量增加了一倍,这是其优势所在。我们在欧洲血统的无关参与者中鉴定了编码蛋白的变异,并进行了罕见变异关联分析,包括 1647 例 IPAH 病例和 18819 例对照。我们还分析了 124 例儿科三联体的新生变异,这些三联体富集了 IPAH 和 APAH-CHD。
有七个具有罕见有害变异的基因与 IPAH 相关,假发现率小于 0.1:三个已知基因(BMPR2、GDF2 和 TBX4),两个最近确定的候选基因(SOX17、KDR),以及两个新的候选基因(纤连蛋白 2、FBLN2;血小板衍生生长因子 D、PDGFD)。新基因仅基于罕见的有害错义变异而被鉴定,而这一变异类型在任何一个队列中都无法充分评估。候选基因在肺和心脏中的表达模式与已知的 PAH 风险基因相似,大多数变异发生在保守的蛋白结构域中。对于儿科 PAH,与背景突变率相比,预测的有害新生变异有显著的负担(2.45×,p=2.5e-5)。至少有八个携带新生变异的新儿科候选基因在肺/心脏发育中具有合理的作用。
对一个大型国际联盟的罕见变异分析确定了两个新的候选基因-FBLN2 和 PDGFD。新基因在血管生成和重塑中具有已知的功能。三联体分析预测,约 15%的儿科 IPAH 可能由新生变异解释。