Rai Shesh N, Qian Chen, Pan Jianmin, Rai Jayesh P, Song Ming, Bagaitkar Juhi, Merchant Michael, Cave Matthew, Egilmez Nejat K, McClain Craig J
Biostatistics and Bioinformatics Facility, James Graham Brown Cancer Center, University of Louisville, Louisville, KY, 40202, USA.
Department of Biostatistics and Bioinformatics, University of Louisville, Louisville, KY, 40202, USA.
Genes Dis. 2019 Dec 24;8(2):215-223. doi: 10.1016/j.gendis.2019.12.005. eCollection 2021 Mar.
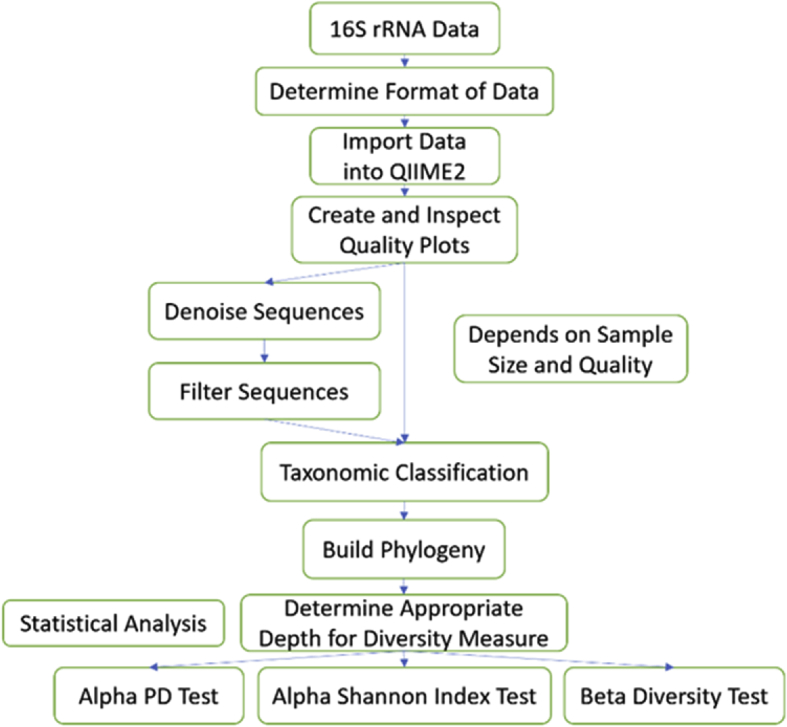
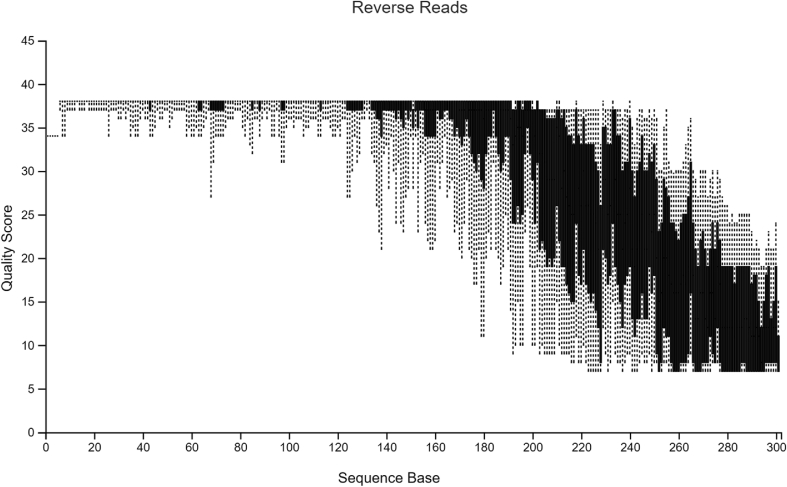
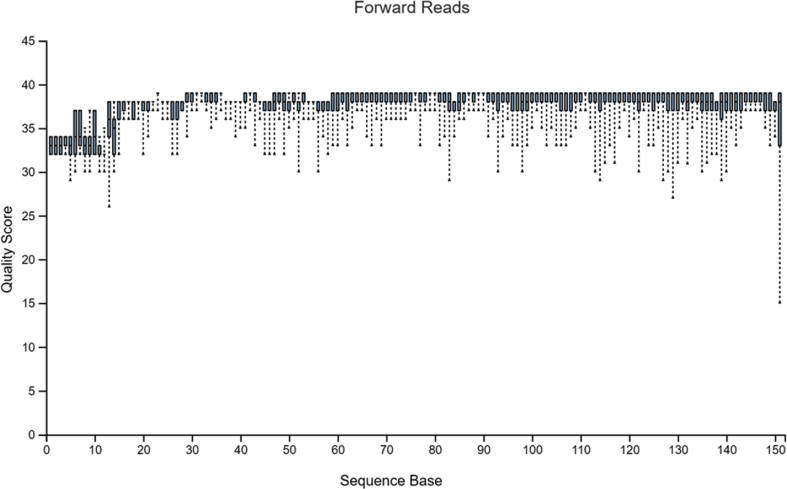
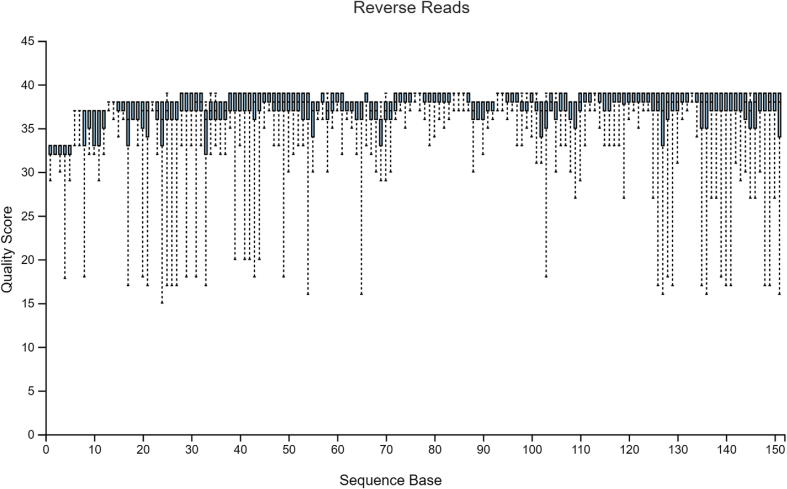
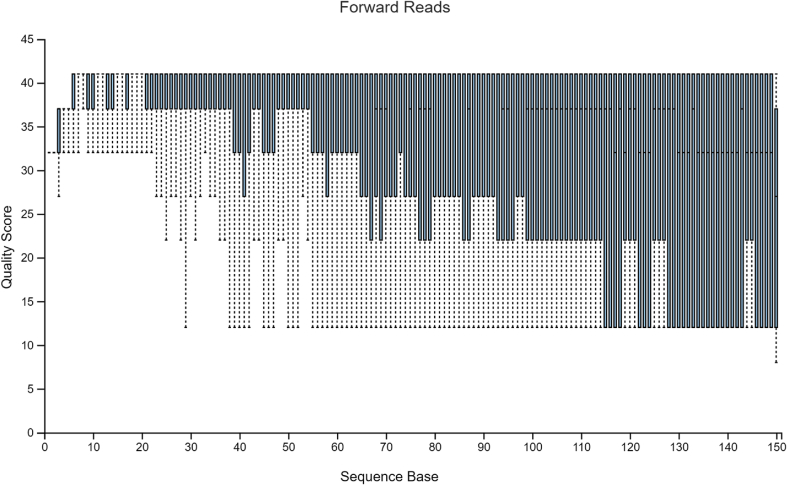
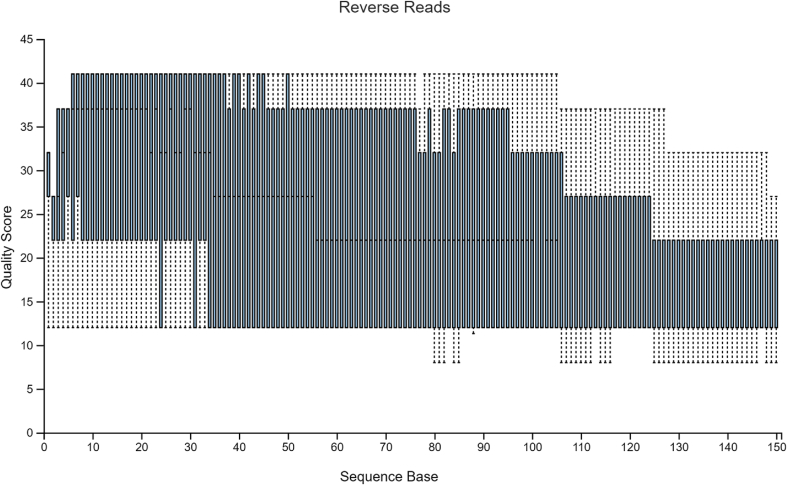
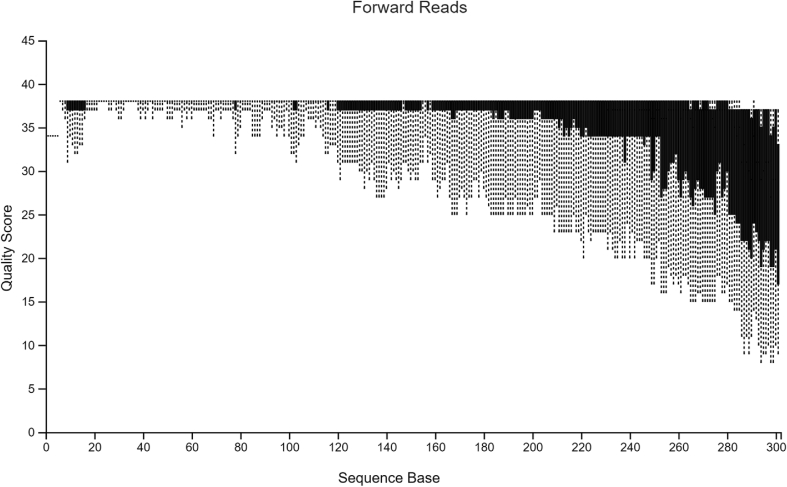
Diversity analysis and taxonomic profiles can be generated from marker-gene sequence data with the help of many available computational tools. The Quantitative Insights into Microbial Ecology Version 2 (QIIME2) has been widely used for 16S rRNA data analysis. While many articles have demonstrated the use of QIIME2 with suitable datasets, the application to pre-clinical data has rarely been talked about. The issues involved in the pre-clinical data include the low-quality score and small sample size that should be addressed properly during analysis. In addition, there are few articles that discuss the detailed statistical methods behind those alpha and beta diversity significance tests that researchers are eager to find. Running the program without knowing the logic behind it is extremely risky. In this article, we first provide a guideline for analyzing 16S rRNA data using QIIME2. Then we will talk about issues in pre-clinical data, and how they could impact the outcome. Finally, we provide brief explanations of statistical methods such as group significance tests and sample size calculation.
借助许多可用的计算工具,可以从标记基因序列数据中生成多样性分析和分类概况。微生物生态学定量见解第2版(QIIME2)已被广泛用于16S rRNA数据分析。虽然许多文章展示了QIIME2在合适数据集上的应用,但很少有人谈论其在临床前数据中的应用。临床前数据涉及的问题包括质量得分低和样本量小,在分析过程中应妥善处理。此外,很少有文章讨论研究人员急于了解的那些α和β多样性显著性检验背后的详细统计方法。在不了解程序逻辑的情况下运行程序风险极大。在本文中,我们首先提供使用QIIME2分析16S rRNA数据的指南。然后我们将讨论临床前数据中的问题,以及它们如何影响结果。最后,我们简要解释诸如组显著性检验和样本量计算等统计方法。