Section on Heritable Disorders of Bone and Extracellular Matrix, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA.
Endocr Rev. 2022 Jan 12;43(1):61-90. doi: 10.1210/endrev/bnab017.
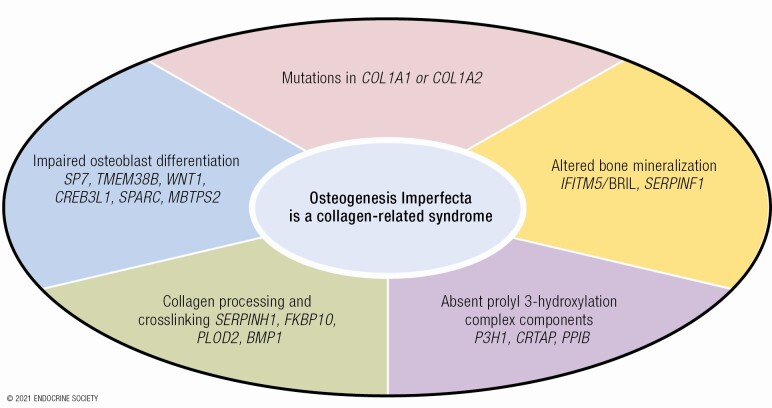
Osteogenesis imperfecta (OI) is a phenotypically and genetically heterogeneous skeletal dysplasia characterized by bone fragility, growth deficiency, and skeletal deformity. Previously known to be caused by defects in type I collagen, the major protein of extracellular matrix, it is now also understood to be a collagen-related disorder caused by defects in collagen folding, posttranslational modification and processing, bone mineralization, and osteoblast differentiation, with inheritance of OI types spanning autosomal dominant and recessive as well as X-linked recessive. This review provides the latest updates on OI, encompassing both classical OI and rare forms, their mechanism, and the signaling pathways involved in their pathophysiology. There is a special emphasis on mutations in type I procollagen C-propeptide structure and processing, the later causing OI with strikingly high bone mass. Types V and VI OI, while notably different, are shown to be interrelated by the interferon-induced transmembrane protein 5 p.S40L mutation that reveals the connection between the bone-restricted interferon-induced transmembrane protein-like protein and pigment epithelium-derived factor pathways. The function of regulated intramembrane proteolysis has been extended beyond cholesterol metabolism to bone formation by defects in regulated membrane proteolysis components site-2 protease and old astrocyte specifically induced-substance. Several recently proposed candidate genes for new types of OI are also presented. Discoveries of new OI genes add complexity to already-challenging OI management; current and potential approaches are summarized.
成骨不全症(OI)是一种表型和遗传异质性的骨骼发育不良,其特征为骨骼脆弱、生长缺陷和骨骼畸形。以前被认为是由Ⅰ型胶原缺陷引起的,Ⅰ型胶原是细胞外基质的主要蛋白,现在也被理解为一种胶原相关疾病,由胶原折叠、翻译后修饰和加工、骨矿化和成骨细胞分化缺陷引起,OI 类型的遗传方式包括常染色体显性和隐性以及 X 连锁隐性遗传。本综述提供了 OI 的最新更新信息,包括经典 OI 和罕见形式、其机制以及涉及它们病理生理学的信号通路。特别强调了Ⅰ型前胶原 C 端肽结构和加工中的突变,后者导致骨量明显增加的 OI。V 型和 VI 型 OI 虽然明显不同,但干扰素诱导跨膜蛋白 5 p.S40L 突变显示它们之间存在相关性,该突变揭示了骨限制干扰素诱导跨膜蛋白样蛋白和色素上皮衍生因子途径之间的联系。调节膜内蛋白水解的功能已从胆固醇代谢扩展到成骨作用,这是通过调节膜蛋白水解成分位点 2 蛋白酶和衰老星形细胞特异性诱导物质缺陷引起的。还提出了几种新的 OI 类型的候选基因。新的 OI 基因的发现增加了已经具有挑战性的 OI 管理的复杂性;总结了当前和潜在的方法。