Broadstreet HEOR, 201 - 343 Railway St, Vancouver, BC, V6A 1A4, Canada.
Sarepta Therapeutics, 215 First St, Cambridge, MA, 02142, USA.
Orphanet J Rare Dis. 2021 May 22;16(1):237. doi: 10.1186/s13023-021-01862-w.
Duchenne muscular dystrophy (DMD) is a severe rare progressive inherited neuromuscular disorder, leading to loss of ambulation (LOA) and premature mortality. The standard of care for patients with DMD has been treatment with corticosteroids for the past decade; however a synthesis of contemporary data describing the clinical course of DMD is lacking. The objective was to summarize age at key clinical milestones (loss of ambulation, scoliosis, ventilation, cardiomyopathy, and mortality) in the corticosteroid-treatment-era.
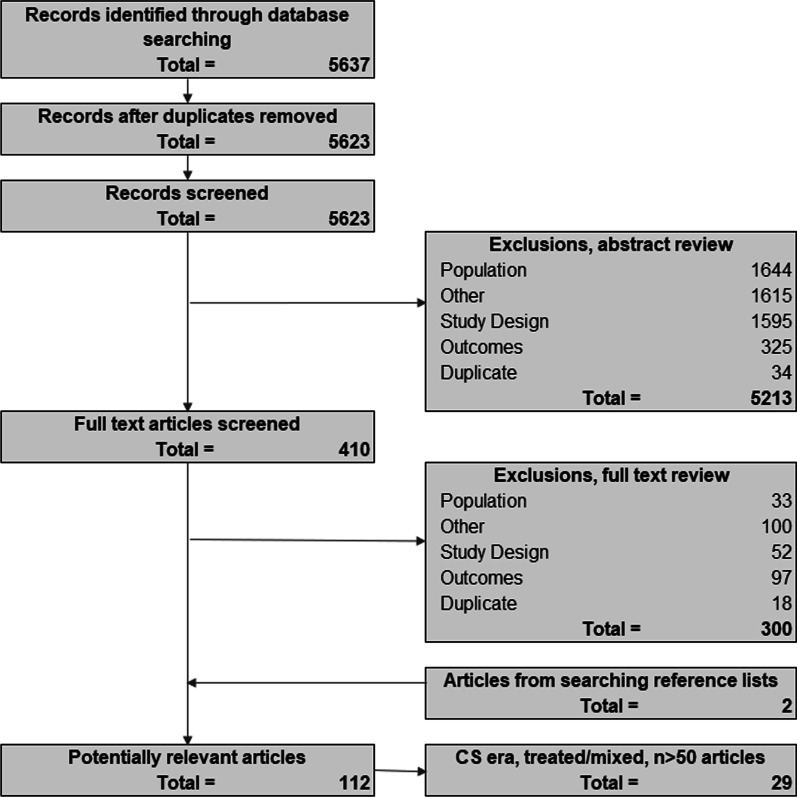
A systematic review was conducted using MEDLINE and EMBASE. The percentage experiencing key clinical milestones, and the mean or median age at those milestones, was synthesized from studies from North American populations, published between 2007 and 2018.
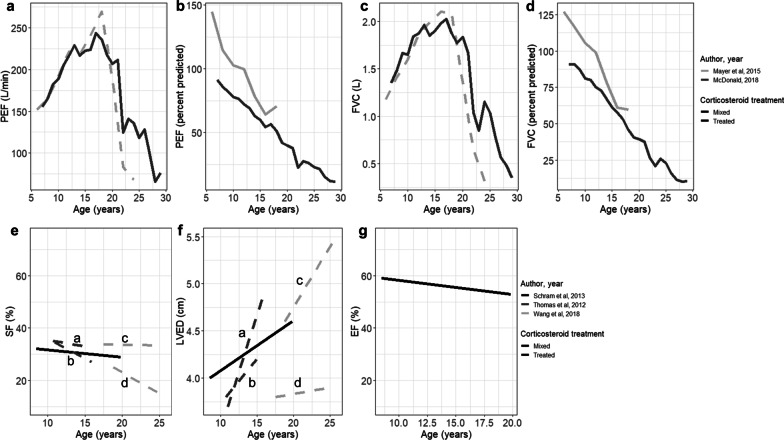
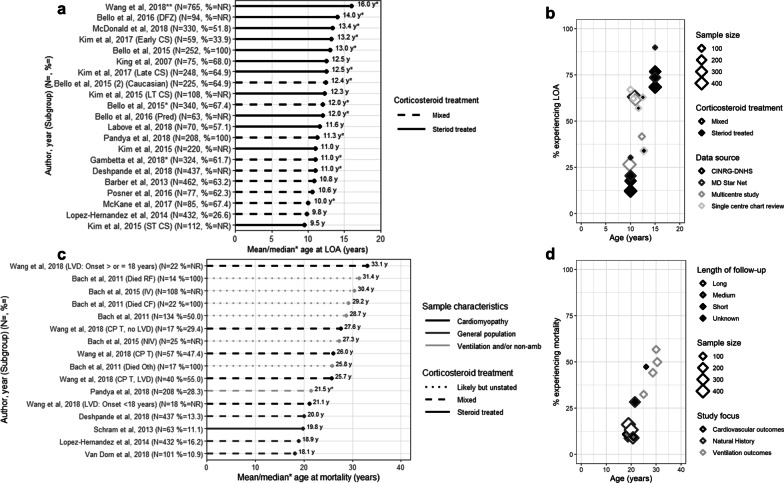
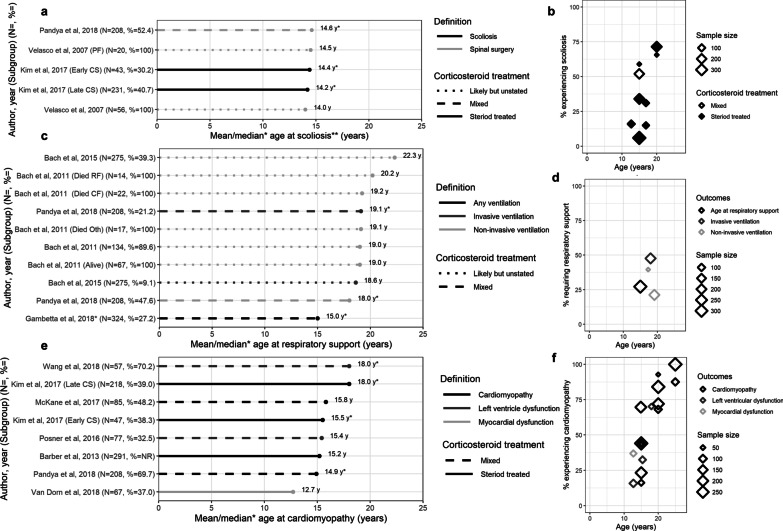
From 5637 abstracts, 29 studies were included. Estimates of the percentage experiencing key clinical milestones, and age at those milestones, showed heterogeneity. Up to 30% of patients lost ambulation by age 10 years, and up to 90% by 15 years of age. The mean age at scoliosis onset was approximately 14 years. Ventilatory support began from 15 to 18 years, and up to half of patients required ventilation by 20 years of age. Registry-based estimates suggest that 70% had evidence of cardiomyopathy by 15 years and almost all by 20 years of age. Finally, mortality rates up to 16% by age 20 years were reported; among those surviving to adulthood mortality was up to 60% by age 30 years.
Contemporary natural history studies from North America report that LOA on average occurs in the early teens, need for ventilation and cardiomyopathy in the late teens, and death in the third or fourth decade of life. Variability in rates may be due to differences in study design, treatment with corticosteroids or other disease-modifying agents, variations in clinical practices, and dystrophin mutations. Despite challenges in synthesizing estimates, these findings help characterize disease progression among contemporary North American DMD patients.
杜氏肌营养不良症(DMD)是一种严重的罕见进行性遗传性神经肌肉疾病,导致行走能力丧失(LOA)和过早死亡。在过去十年中,DMD 患者的标准治疗方法一直是使用皮质类固醇治疗;然而,缺乏对描述 DMD 临床过程的当代数据的综合分析。本研究旨在总结皮质类固醇治疗时代 DMD 患者在关键临床里程碑(丧失行走能力、脊柱侧凸、通气、心肌病和死亡)的年龄。
使用 MEDLINE 和 EMBASE 进行系统综述。从北美人群发表的 2007 年至 2018 年的研究中综合了关键临床里程碑的发生率百分比以及达到这些里程碑的平均或中位数年龄。
从 5637 篇摘要中,纳入了 29 项研究。关键临床里程碑的发生率百分比以及达到这些里程碑的年龄的估计值存在异质性。多达 30%的患者在 10 岁之前丧失行走能力,多达 90%的患者在 15 岁之前丧失行走能力。脊柱侧凸发病的平均年龄约为 14 岁。通气支持从 15 岁到 18 岁开始,多达一半的患者在 20 岁之前需要通气。基于登记的估计表明,70%的患者在 15 岁时出现心肌病证据,几乎所有患者在 20 岁时都出现心肌病证据。最后,报告了高达 20 岁时 16%的死亡率;在成年后存活的患者中,30 岁时的死亡率高达 60%。
来自北美的当代自然史研究报告称,LOA 平均发生在青少年早期,通气和心肌病发生在青少年晚期,30 多岁时死亡。发生率的差异可能是由于研究设计、皮质类固醇或其他疾病修饰剂的治疗、临床实践的差异以及肌营养不良蛋白基因突变的差异。尽管在综合评估方面存在挑战,但这些发现有助于描述当代北美 DMD 患者的疾病进展。