González-Barriga Anchel, Lallemant Louison, Dincã Diana M, Braz Sandra O, Polvèche Hélène, Magneron Paul, Pionneau Cédric, Huguet-Lachon Aline, Claude Jean-Baptiste, Chhuon Cerina, Guerrera Ida Chiara, Bourgeois Cyril F, Auboeuf Didier, Gourdon Geneviève, Gomes-Pereira Mário
Sorbonne Université, Inserm, Institut de Myologie, Centre de Recherche en Myologie, Paris, France.
Inserm UMR 1163, Institut Imagine, Université Paris Cité, Paris, France.
Front Cell Neurosci. 2021 May 5;15:662035. doi: 10.3389/fncel.2021.662035. eCollection 2021.
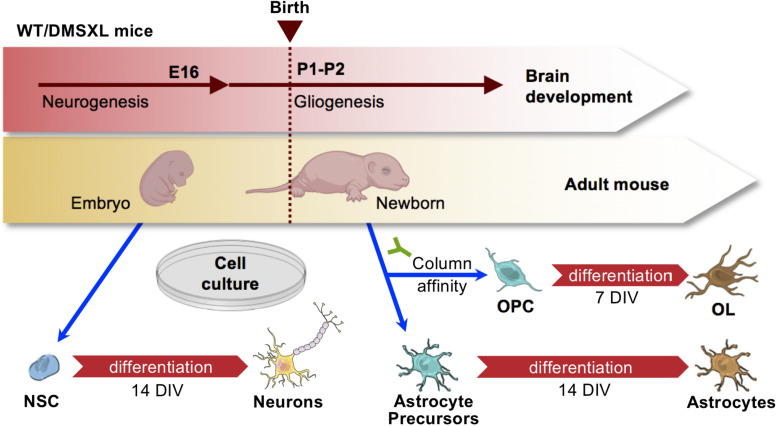
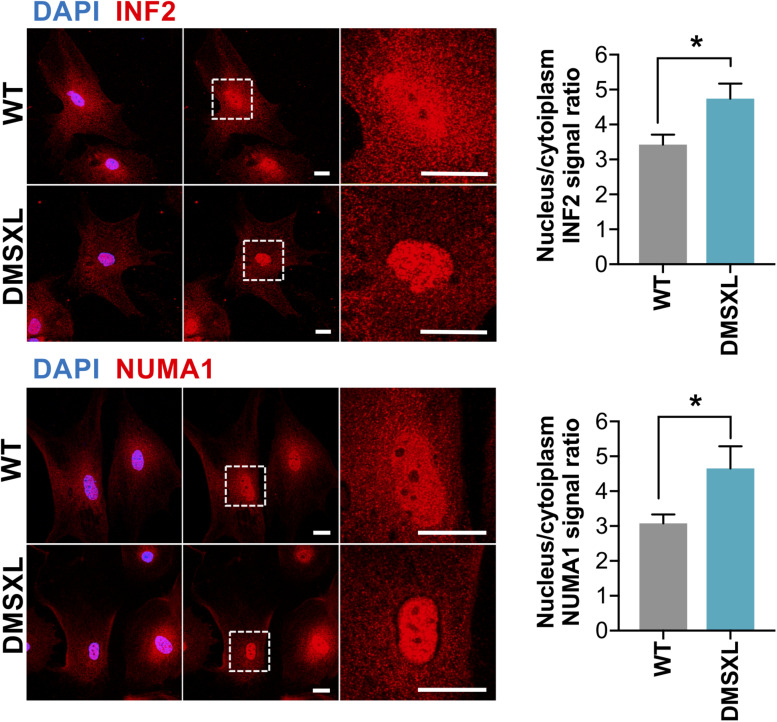
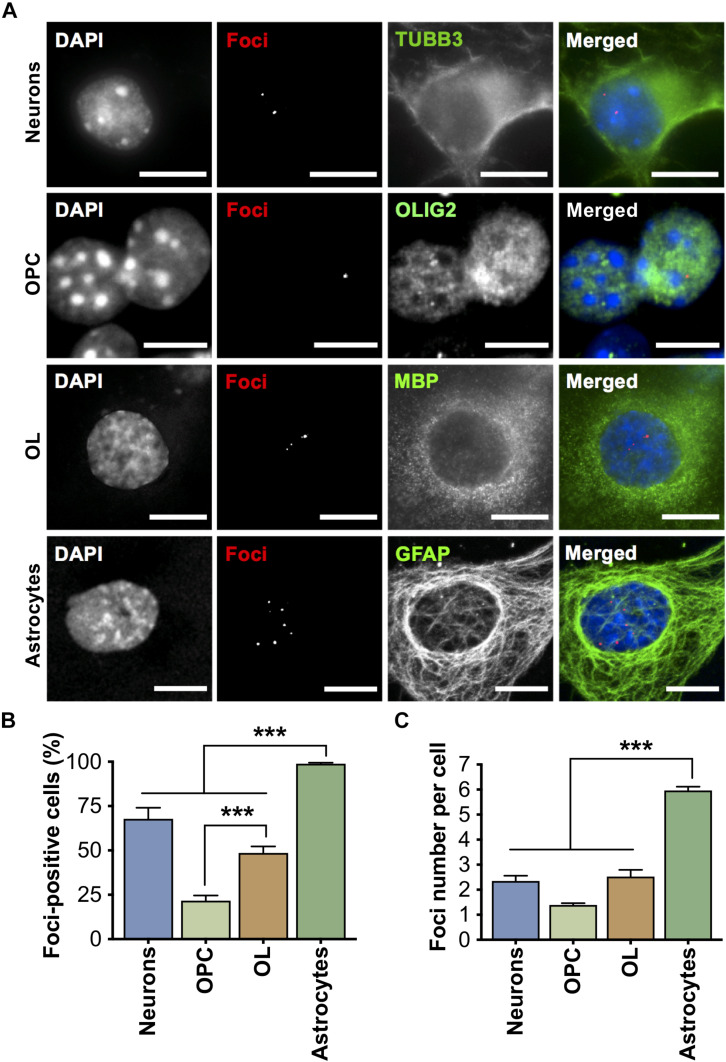
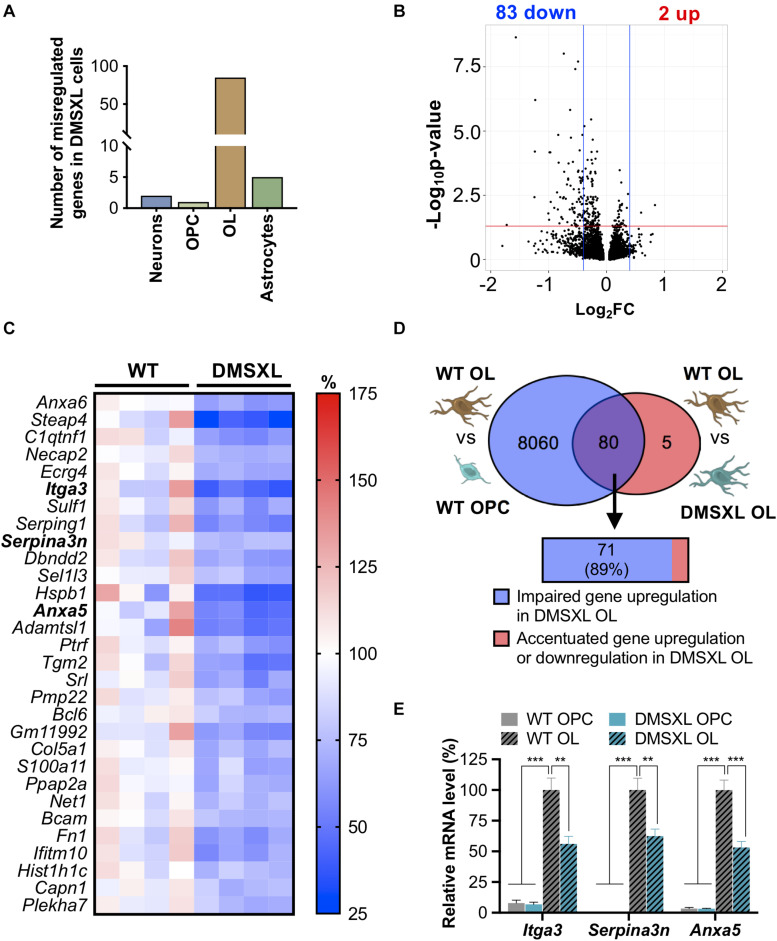
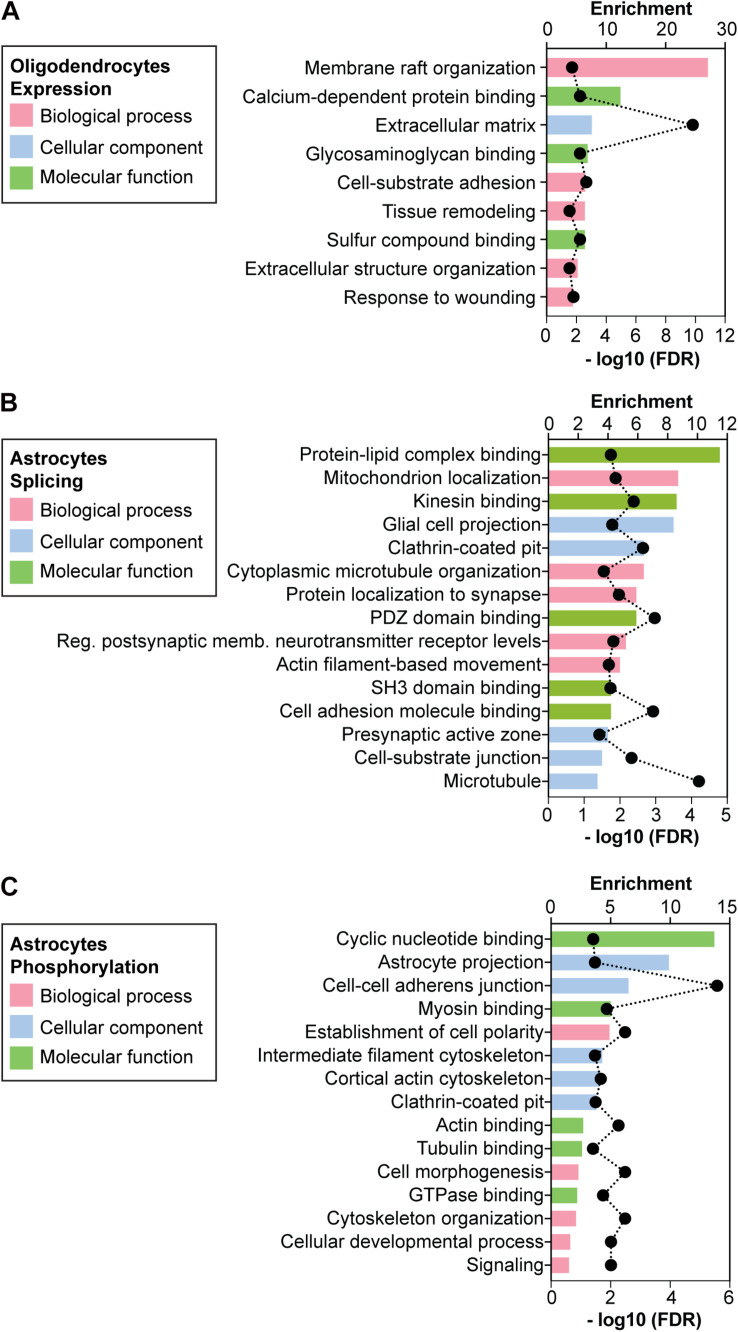
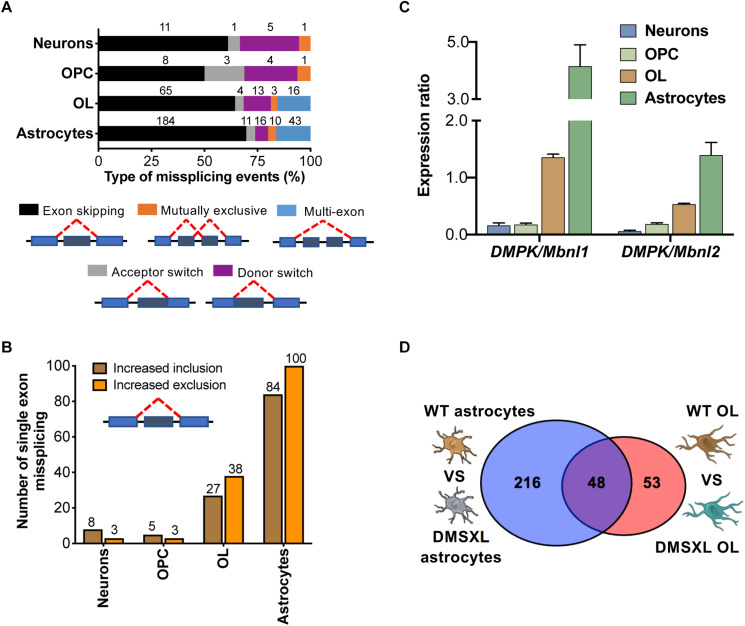
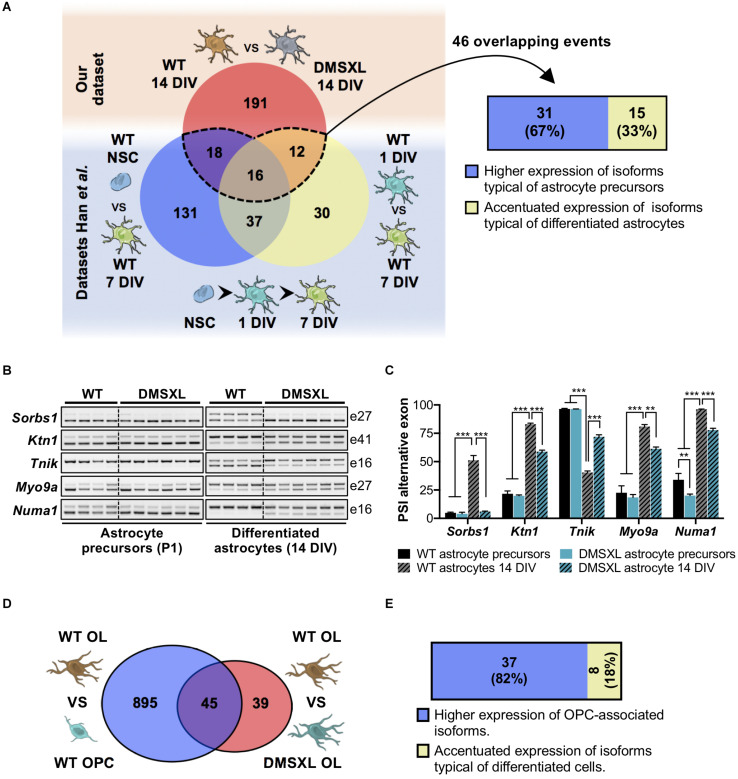
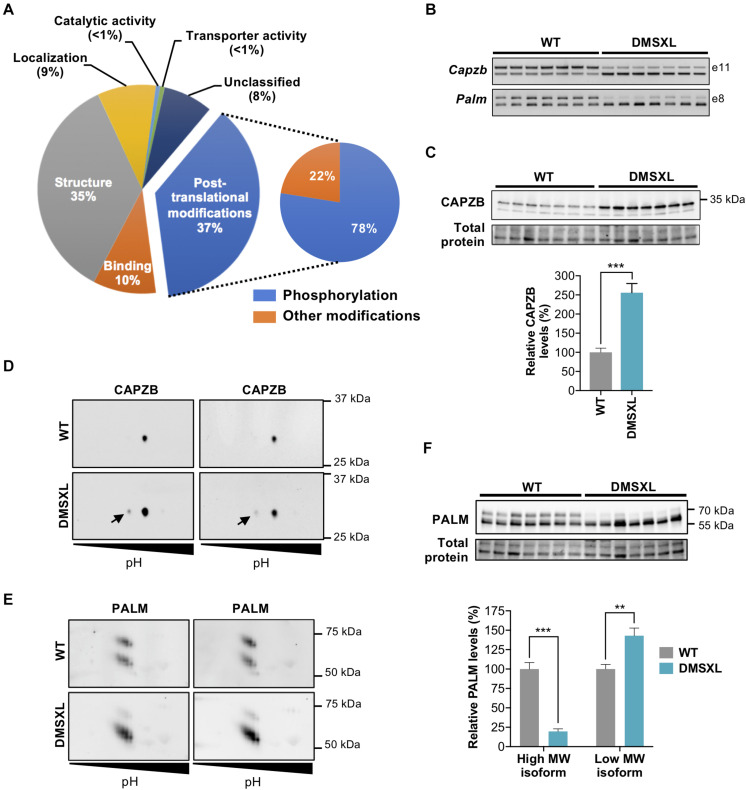
Myotonic dystrophy type 1 (DM1) is a neuromuscular disorder caused by a non-coding CTG repeat expansion in the gene. This mutation generates a toxic CUG RNA that interferes with the RNA processing of target genes in multiple tissues. Despite debilitating neurological impairment, the pathophysiological cascade of molecular and cellular events in the central nervous system (CNS) has been less extensively characterized than the molecular pathogenesis of muscle/cardiac dysfunction. Particularly, the contribution of different cell types to DM1 brain disease is not clearly understood. We first used transcriptomics to compare the impact of expanded CUG RNA on the transcriptome of primary neurons, astrocytes and oligodendrocytes derived from DMSXL mice, a transgenic model of DM1. RNA sequencing revealed more frequent expression and splicing changes in glia than neuronal cells. In particular, primary DMSXL oligodendrocytes showed the highest number of transcripts differentially expressed, while DMSXL astrocytes displayed the most severe splicing dysregulation. Interestingly, the expression and splicing defects of DMSXL glia recreated molecular signatures suggestive of impaired cell differentiation: while DMSXL oligodendrocytes failed to upregulate a subset of genes that are naturally activated during the oligodendroglia differentiation, a significant proportion of missplicing events in DMSXL oligodendrocytes and astrocytes increased the expression of RNA isoforms typical of precursor cell stages. Together these data suggest that expanded CUG RNA in glial cells affects preferentially differentiation-regulated molecular events. This hypothesis was corroborated by gene ontology (GO) analyses, which revealed an enrichment for biological processes and cellular components with critical roles during cell differentiation. Finally, we combined exon ontology with phosphoproteomics and cell imaging to explore the functional impact of CUG-associated spliceopathy on downstream protein metabolism. Changes in phosphorylation, protein isoform expression and intracellular localization in DMSXL astrocytes demonstrate the far-reaching impact of the DM1 repeat expansion on cell metabolism. Our multi-omics approaches provide insight into the mechanisms of CUG RNA toxicity in the CNS with cell type resolution, and support the priority for future research on non-neuronal mechanisms and proteomic changes in DM1 brain disease.
1型强直性肌营养不良(DM1)是一种由该基因中非编码CTG重复序列扩增引起的神经肌肉疾病。这种突变产生一种有毒的CUG RNA,它会干扰多个组织中靶基因的RNA加工过程。尽管存在使人衰弱的神经功能障碍,但与肌肉/心脏功能障碍的分子发病机制相比,中枢神经系统(CNS)中分子和细胞事件的病理生理级联反应尚未得到广泛的表征。特别是,不同细胞类型对DM1脑疾病的贡献尚不清楚。我们首先使用转录组学来比较扩增的CUG RNA对源自DMSXL小鼠(一种DM1转基因模型)的原代神经元、星形胶质细胞和少突胶质细胞转录组的影响。RNA测序显示,与神经元细胞相比,神经胶质细胞中表达和剪接变化更频繁。特别是,原代DMSXL少突胶质细胞显示出差异表达转录本数量最多,而DMSXL星形胶质细胞则表现出最严重的剪接失调。有趣的是,DMSXL神经胶质细胞的表达和剪接缺陷重现了提示细胞分化受损的分子特征:虽然DMSXL少突胶质细胞未能上调少突胶质细胞分化过程中自然激活的一部分基因,但DMSXL少突胶质细胞和星形胶质细胞中相当一部分错配剪接事件增加了前体细胞阶段典型RNA异构体的表达。这些数据共同表明,神经胶质细胞中扩增的CUG RNA优先影响分化调节的分子事件。基因本体(GO)分析证实了这一假设,该分析揭示了在细胞分化过程中起关键作用的生物过程和细胞成分的富集。最后,我们将外显子本体与磷酸蛋白质组学和细胞成像相结合,以探索CUG相关剪接病对下游蛋白质代谢的功能影响。DMSXL星形胶质细胞中磷酸化、蛋白质异构体表达和细胞内定位的变化证明了DM1重复序列扩增对细胞代谢的深远影响。我们的多组学方法以细胞类型分辨率深入了解了CUG RNA在中枢神经系统中的毒性机制,并支持未来对DM1脑疾病中非神经元机制和蛋白质组变化进行研究的优先性。