Liu Zhaoyan, Zhu Wei, Gnatenko Dmitri V, Nesbitt Natasha M, Bahou Wadie F
Department of Applied Mathematics and Statistics, Stony Brook University, Stony Brook, NY 11794 (USA).
Department of Medicine, Stony Brook University, Stony Brook, NY 11794 (USA).
Data Brief. 2021 Apr 22;36:107080. doi: 10.1016/j.dib.2021.107080. eCollection 2021 Jun.
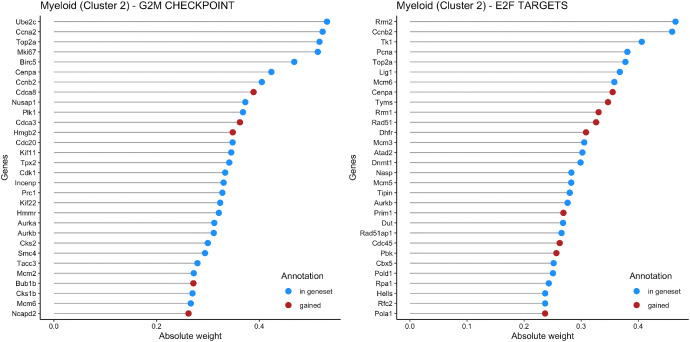
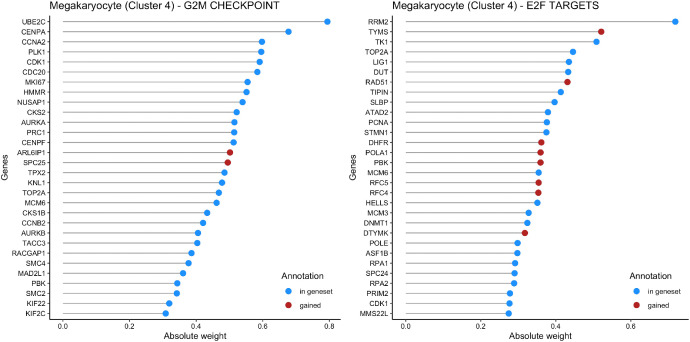
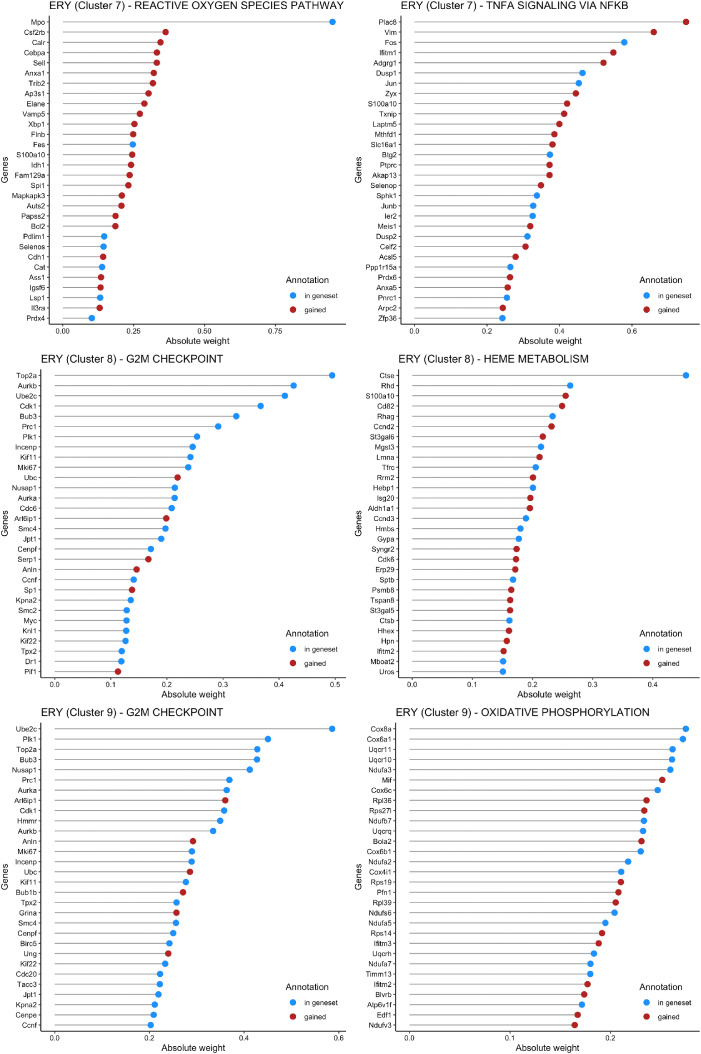
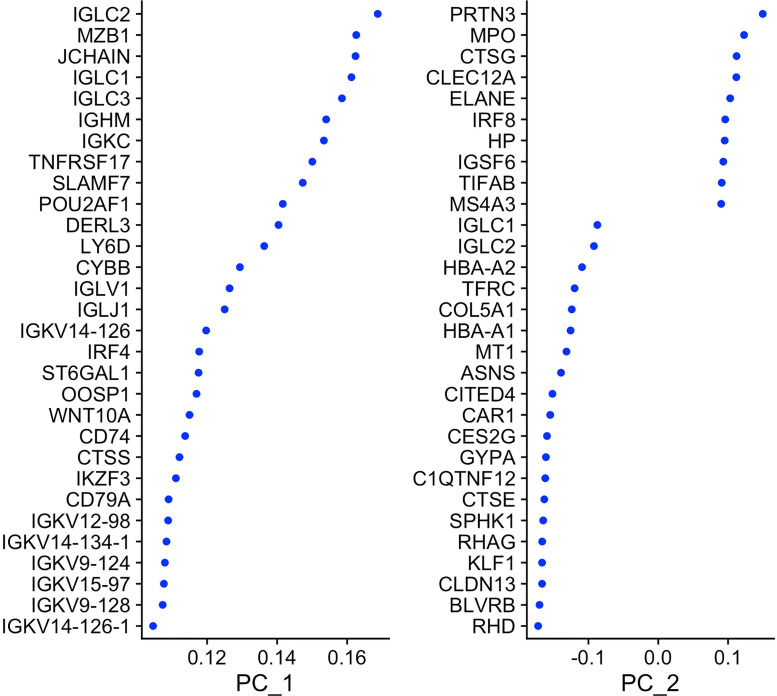
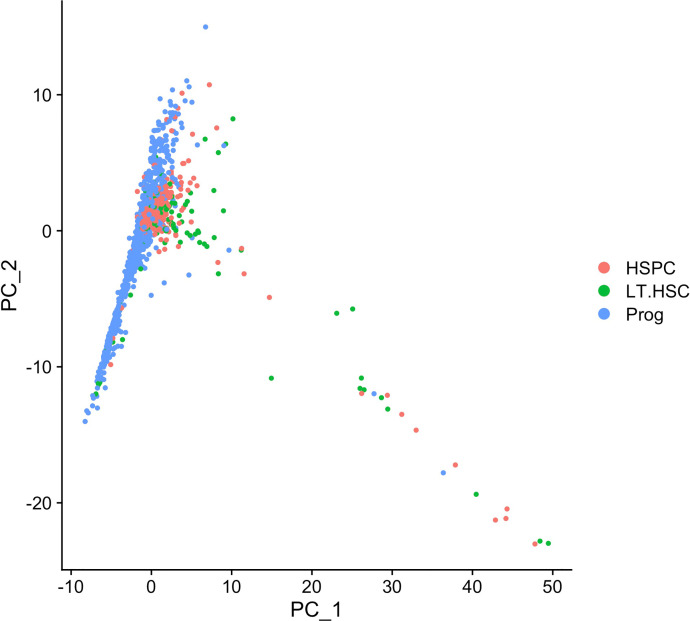
Genetic pathways regulating hematopoietic lineage commitment at critical stages of development remain incompletely characterized. To better delineate genetic sources of variability regulating cellular speciation during steady-state hematopoiesis, we applied a factorial single-cell latent variable model (f-scLVM) to decompose single-cell transcriptome heterogeneity into interpretable biological factors (refined pathway annotations or gene sets without annotation) dynamically regulating cell fate. Hematopoietic single cell transcriptomic raw sequencing data extracted from 1,920 hematopoietic stem and progenitor cells (HSPCs) derived from 12-week-old female mice were used for data analysis and model development. These single cell RNA sequencing data were subsequently analyzed using the factorial single-cell latent variable model (f-scLVM), with their heterogeneity decomposed into interpretable biological factors. The top biological factors underlying the basal hematopoiesis were subsequently identified for the aggregate, and lineage-restricted (myeloid, megakaryocyte, erythroid) progenitor cells. For a subset of factors, data were independently verified experimentally in a companion research paper [1]. These data facilitate the identification of novel subpopulations and adjust gene sets to discover new marker genes and hidden confounding factors driving basal hematopoiesis.
在发育的关键阶段调节造血谱系定向的遗传途径仍未完全明确。为了更好地描绘在稳态造血过程中调节细胞分化的遗传变异来源,我们应用了一种因子单细胞潜在变量模型(f-scLVM),将单细胞转录组异质性分解为动态调节细胞命运的可解释生物学因子(精细的通路注释或无注释的基因集)。从12周龄雌性小鼠来源的1920个造血干细胞和祖细胞(HSPCs)中提取的造血单细胞转录组原始测序数据用于数据分析和模型开发。随后使用因子单细胞潜在变量模型(f-scLVM)对这些单细胞RNA测序数据进行分析,将其异质性分解为可解释的生物学因子。随后确定了基础造血、聚集以及谱系受限(髓系、巨核细胞、红系)祖细胞的首要生物学因子。对于一部分因子,数据在一篇配套研究论文[1]中通过实验进行了独立验证。这些数据有助于识别新的亚群并调整基因集,以发现驱动基础造血的新标记基因和隐藏的混杂因素。