Zhao Xin, Gao Shouguo, Kajigaya Sachiko, Liu Qingguo, Wu Zhijie, Feng Xingmin, Zhang Fengkui, Young Neal S
Hematology Branch, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD, 20892, USA.
State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, 300020, China.
BMC Res Notes. 2020 Nov 10;13(1):514. doi: 10.1186/s13104-020-05357-y.
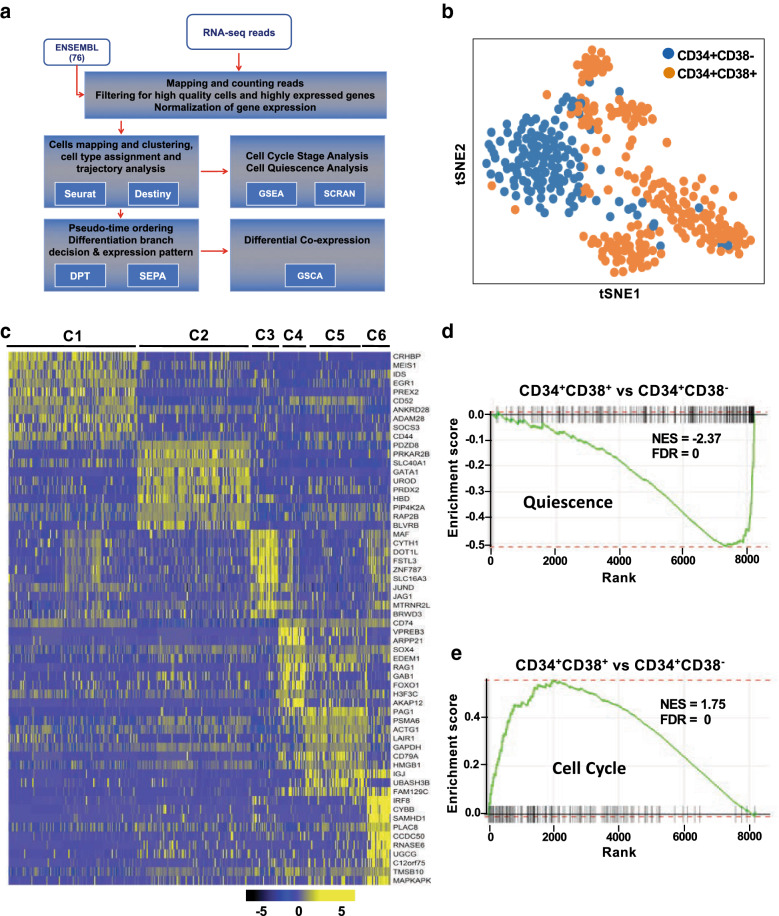
Single cell methodology enables detection and quantification of transcriptional changes and unravelling dynamic aspects of the transcriptional heterogeneity not accessible using bulk sequencing approaches. We have applied single-cell RNA-sequencing (scRNA-seq) to fresh human bone marrow CD34 cells and profiled 391 single hematopoietic stem/progenitor cells (HSPCs) from healthy donors to characterize lineage- and stage-specific transcription during hematopoiesis.
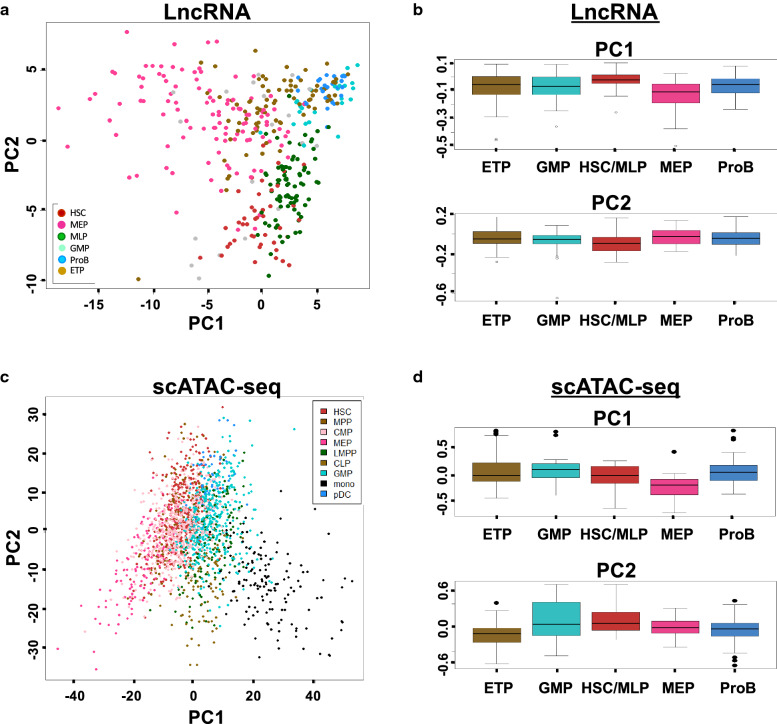
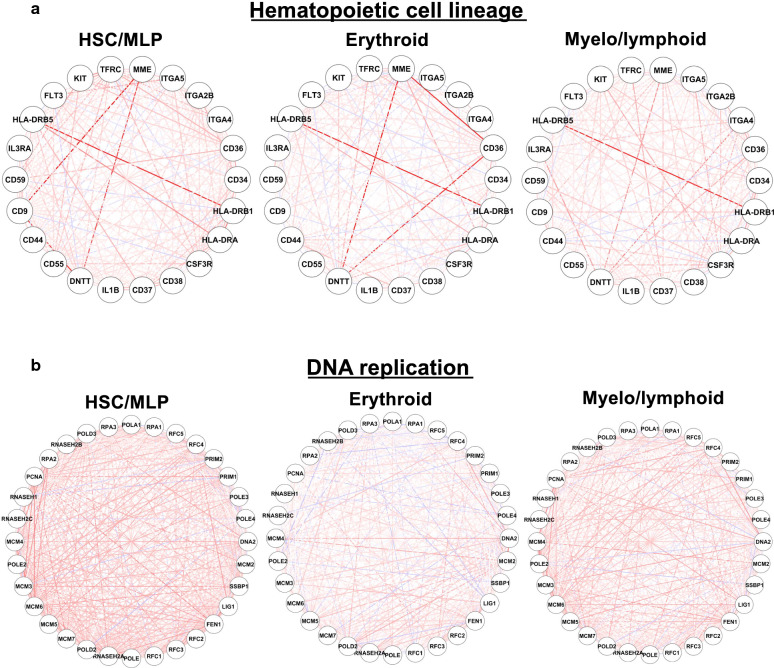
Cells clustered into six distinct groups, which could be assigned to known HSPC subpopulations based on lineage specific genes. Reconstruction of differentiation trajectories in single cells revealed four committed lineages derived from HSCs, as well as dynamic expression changes underlying cell fate during early erythroid-megakaryocytic, lymphoid, and granulocyte-monocyte differentiation. A similar non-hierarchical pattern of hematopoiesis could be derived from analysis of published single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq), consistent with a sequential relationship between chromatin dynamics and regulation of gene expression during lineage commitment (first, altered chromatin conformation, then mRNA transcription). Computationally, we have reconstructed molecular trajectories connecting HSCs directly to four hematopoietic lineages. Integration of long noncoding RNA (lncRNA) expression from the same cells demonstrated mRNA transcriptome, lncRNA, and the epigenome were highly homologous in their pattern of gene activation and suppression during hematopoietic cell differentiation.
单细胞方法能够检测和量化转录变化,并揭示使用批量测序方法无法获得的转录异质性的动态方面。我们已将单细胞RNA测序(scRNA-seq)应用于新鲜的人骨髓CD34细胞,并对来自健康供体的391个单个造血干细胞/祖细胞(HSPC)进行了分析,以表征造血过程中谱系和阶段特异性转录。
细胞聚集成六个不同的组,根据谱系特异性基因可将其分配到已知的HSPC亚群。单细胞中分化轨迹的重建揭示了源自造血干细胞的四个定向谱系,以及早期红系-巨核系、淋巴系和粒细胞-单核细胞分化过程中细胞命运背后的动态表达变化。通过对已发表的转座酶可及染色质测序单细胞分析(scATAC-seq)的分析,也可得出类似的非分层造血模式,这与谱系定向过程中染色质动力学与基因表达调控之间的顺序关系一致(首先,染色质构象改变,然后是mRNA转录)。通过计算,我们重建了将造血干细胞直接连接到四个造血谱系的分子轨迹。对同一细胞的长链非编码RNA(lncRNA)表达进行整合,结果表明mRNA转录组、lncRNA和表观基因组在造血细胞分化过程中的基因激活和抑制模式高度同源。