Crotti Lia, Celano Giuseppe, Dagradi Federica, Schwartz Peter J
Section of Cardiology, Department of Lung, Blood and Heart, University of Pavia, Pavia, Italy.
Orphanet J Rare Dis. 2008 Jul 7;3:18. doi: 10.1186/1750-1172-3-18.
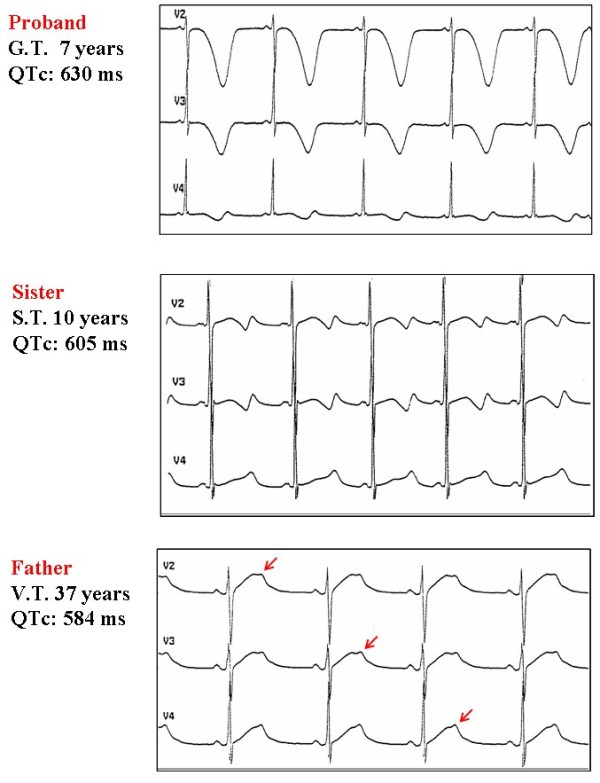
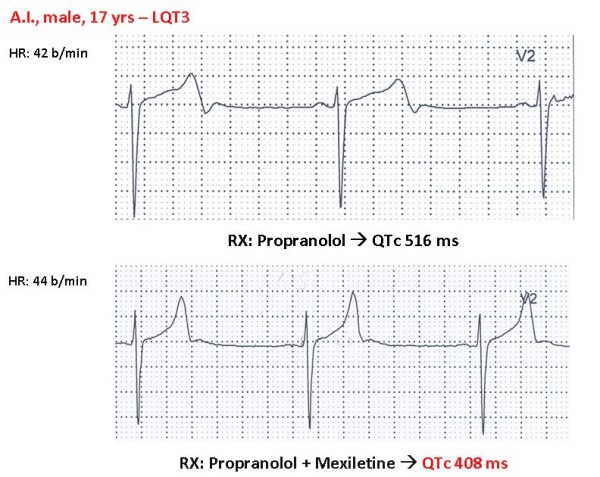
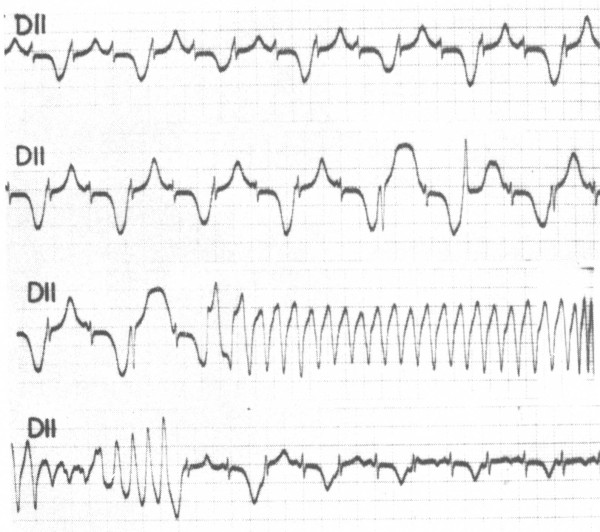
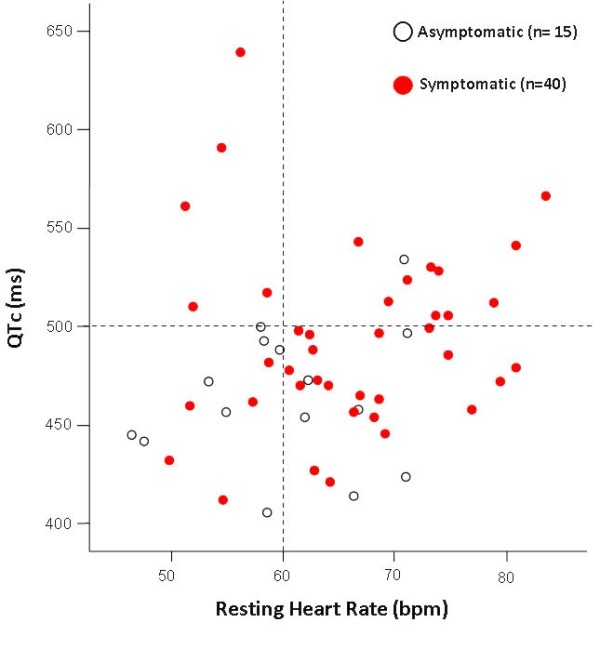
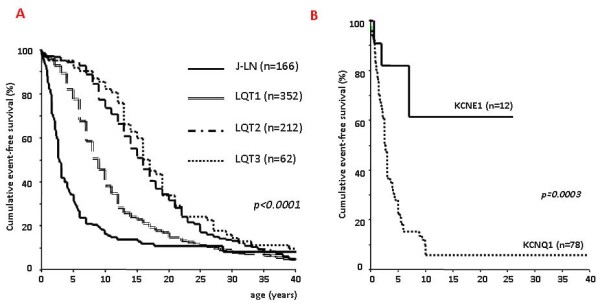
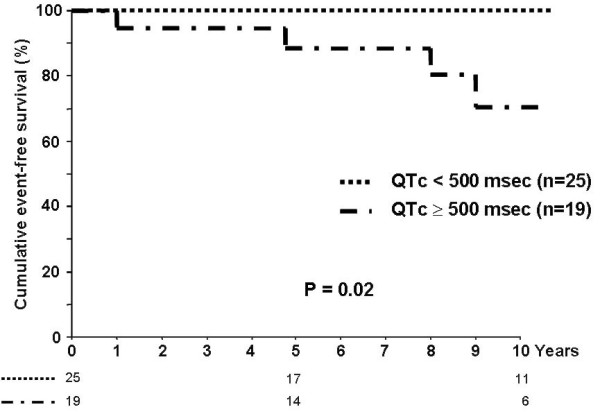
Congenital long QT syndrome (LQTS) is a hereditary cardiac disease characterized by a prolongation of the QT interval at basal ECG and by a high risk of life-threatening arrhythmias. Disease prevalence is estimated at close to 1 in 2,500 live births. The two cardinal manifestations of LQTS are syncopal episodes, that may lead to cardiac arrest and sudden cardiac death, and electrocardiographic abnormalities, including prolongation of the QT interval and T wave abnormalities. The genetic basis of the disease was identified in the mid-nineties and all the LQTS genes identified so far encode cardiac ion channel subunits or proteins involved in modulating ionic currents. Mutations in these genes (KCNQ1, KCNH2, KCNE1, KCNE2, CACNA1c, CAV3, SCN5A, SCN4B) cause the disease by prolonging the duration of the action potential. The most prevalent LQTS variant (LQT1) is caused by mutations in the KCNQ1 gene, with approximately half of the genotyped patients carrying KCNQ1 mutations. Given the characteristic features of LQTS, the typical cases present no diagnostic difficulties for physicians aware of the disease. However, borderline cases are more complex and require the evaluation of various electrocardiographic, clinical, and familial findings, as proposed in specific diagnostic criteria. Additionally, molecular screening is now part of the diagnostic process. Treatment should always begin with beta-blockers, unless there are valid contraindications. If the patient has one more syncope despite a full dose beta-blockade, left cardiac sympathetic denervation (LCSD) should be performed without hesitation and implantable cardioverter defibrillator (ICD) therapy should be considered with the final decision being based on the individual patient characteristics (age, sex, clinical history, genetic subgroup including mutation-specific features in some cases, presence of ECG signs - including 24-hour Holter recordings - indicating high electrical instability). The prognosis of the disease is usually good in patients that are correctly diagnosed and treated. However, there are a few exceptions: patients with Timothy syndrome, patients with Jervell Lange-Nielsen syndrome carrying KCNQ1 mutations and LQT3 patients with 2:1 atrio-ventricular block and very early occurrence of cardiac arrhythmias.
先天性长QT综合征(LQTS)是一种遗传性心脏病,其特征是基础心电图QT间期延长,且有发生危及生命的心律失常的高风险。该病的患病率估计接近每2500例活产中有1例。LQTS的两个主要表现是晕厥发作,这可能导致心脏骤停和心源性猝死,以及心电图异常,包括QT间期延长和T波异常。该疾病的遗传基础在20世纪90年代中期被确定,迄今为止所有已确定的LQTS基因都编码心脏离子通道亚基或参与调节离子电流的蛋白质。这些基因(KCNQ1、KCNH2、KCNE1、KCNE2、CACNA1c、CAV3、SCN5A、SCN4B)中的突变通过延长动作电位的持续时间导致疾病。最常见的LQTS变异型(LQT1)是由KCNQ1基因突变引起的,大约一半的基因分型患者携带KCNQ1突变。鉴于LQTS的特征,典型病例对于了解该疾病的医生来说不存在诊断困难。然而,临界病例更为复杂,需要根据特定诊断标准评估各种心电图、临床和家族性发现。此外,分子筛查现在是诊断过程的一部分。治疗应始终从β受体阻滞剂开始,除非有有效的禁忌症。如果患者在足量β受体阻滞剂治疗下仍有一次或多次晕厥,应毫不犹豫地进行左侧心脏交感神经去神经术(LCSD),并应考虑植入式心脏复律除颤器(ICD)治疗,最终决策应基于个体患者特征(年龄、性别、临床病史、遗传亚组,在某些情况下包括特定突变特征、心电图征象的存在——包括24小时动态心电图记录——表明电不稳定性高)。该疾病在正确诊断和治疗的患者中预后通常良好。然而,也有一些例外情况:患有蒂莫西综合征的患者、携带KCNQ1突变的耶尔韦尔-朗格-尼尔森综合征患者以及患有2:1房室传导阻滞且心律失常很早发生的LQT3患者。