Mass Spectrometry Hub, University of Auckland, Auckland, New Zealand.
School of Biological Sciences, University of Auckland, Auckland, New Zealand.
Nat Commun. 2021 May 28;12(1):3241. doi: 10.1038/s41467-021-23461-w.
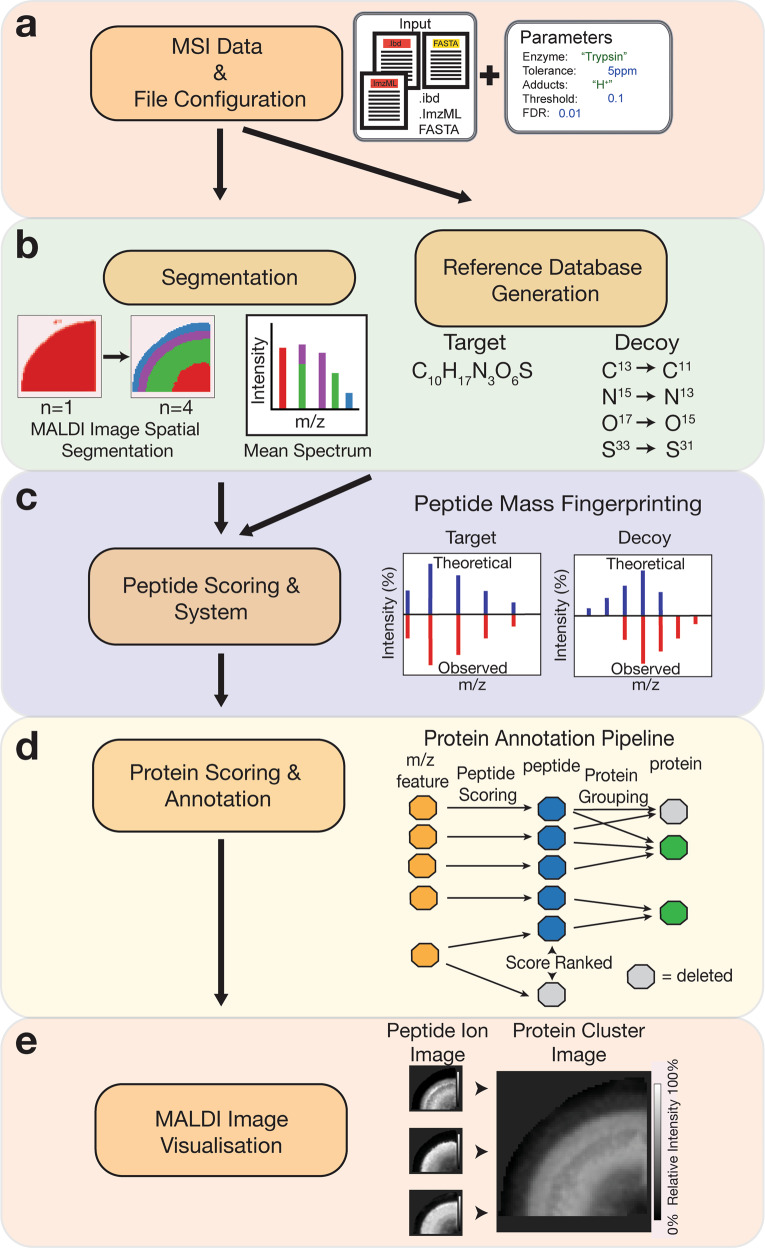
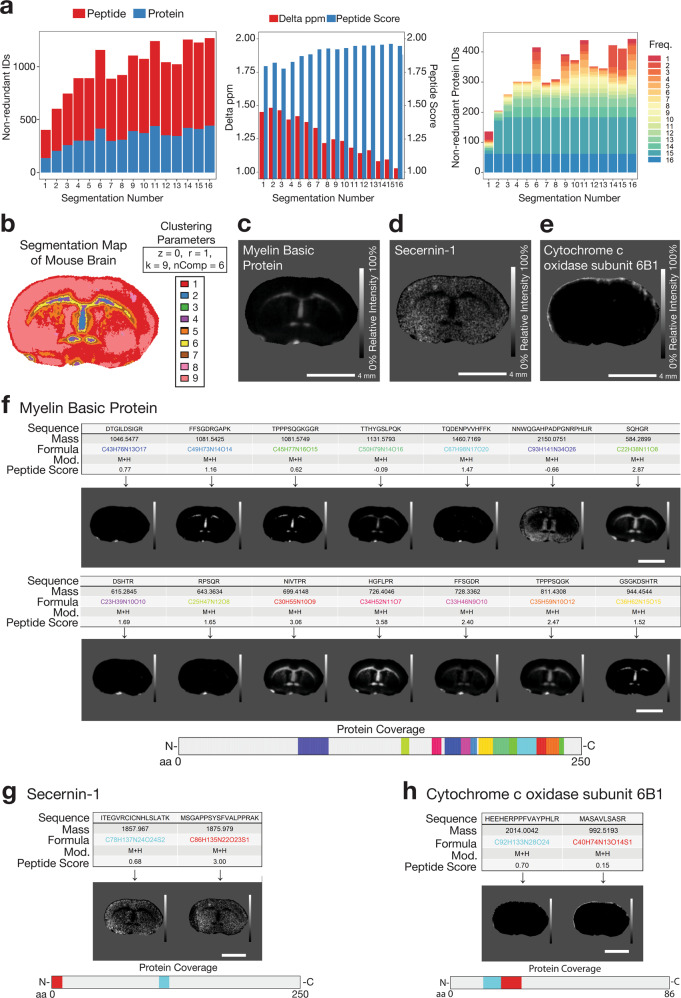
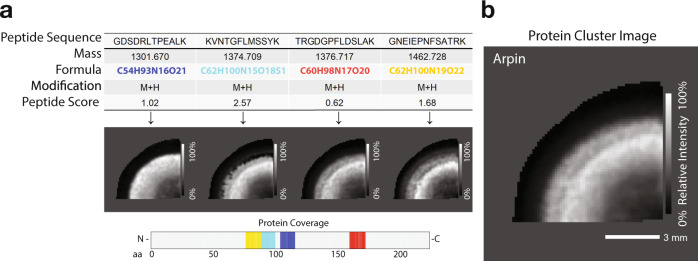
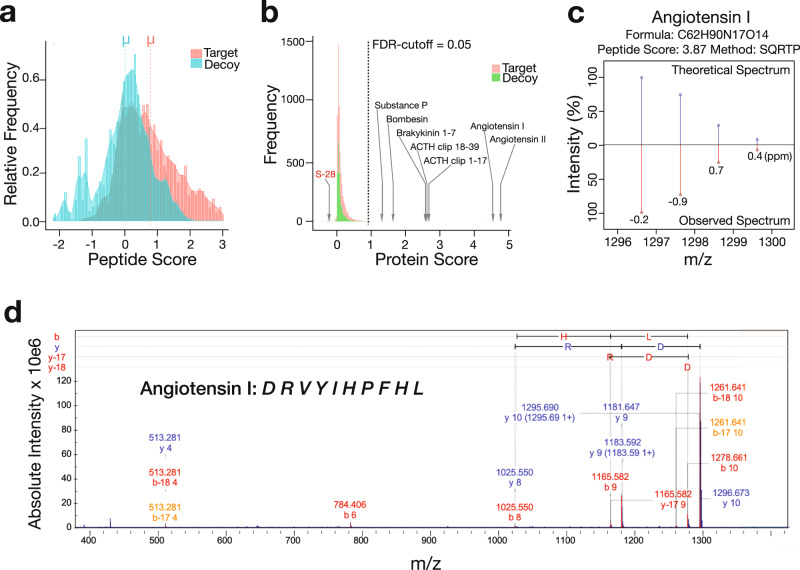
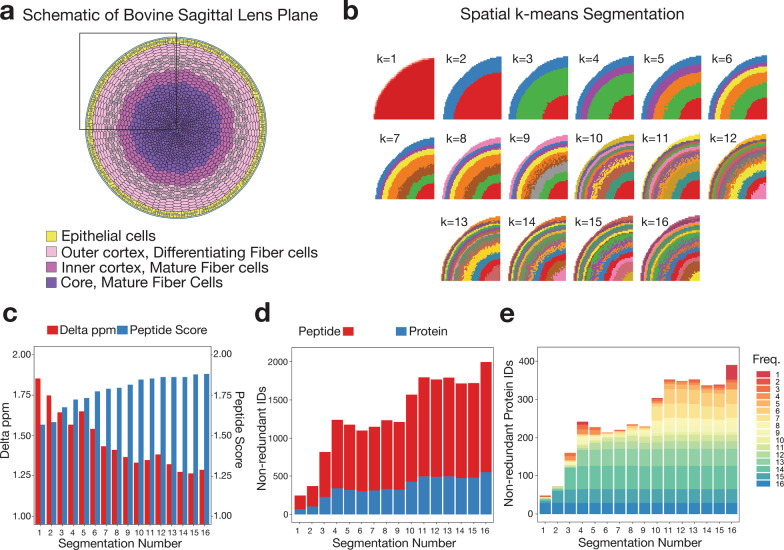
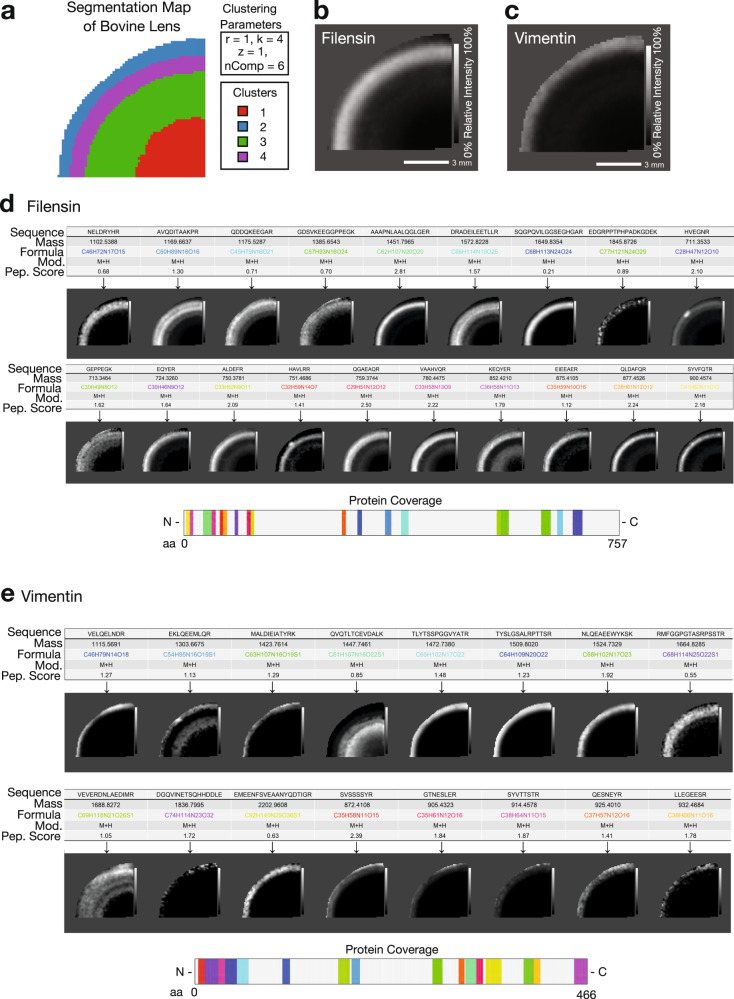
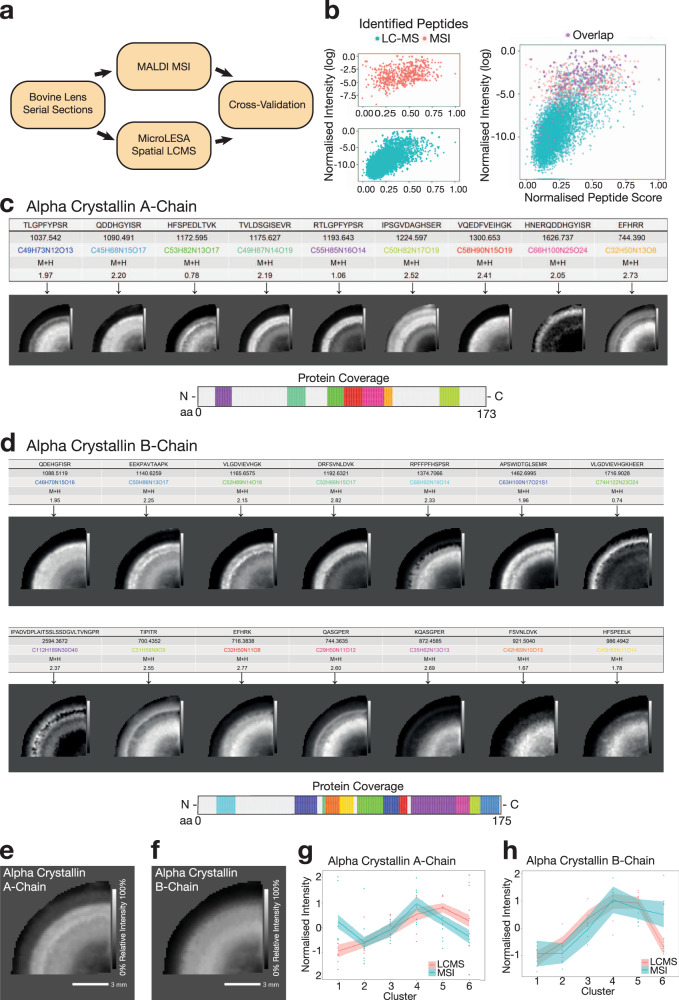
Spatial proteomics has the potential to significantly advance our understanding of biology, physiology and medicine. Matrix-assisted laser desorption/ionisation mass spectrometry imaging (MALDI-MSI) is a powerful tool in the spatial proteomics field, enabling direct detection and registration of protein abundance and distribution across tissues. MALDI-MSI preserves spatial distribution and histology allowing unbiased analysis of complex, heterogeneous tissues. However, MALDI-MSI faces the challenge of simultaneous peptide quantification and identification. To overcome this, we develop and validate HIT-MAP (High-resolution Informatics Toolbox in MALDI-MSI Proteomics), an open-source bioinformatics workflow using peptide mass fingerprint analysis and a dual scoring system to computationally assign peptide and protein annotations to high mass resolution MSI datasets and generate customisable spatial distribution maps. HIT-MAP will be a valuable resource for the spatial proteomics community for analysing newly generated and retrospective datasets, enabling robust peptide and protein annotation and visualisation in a wide array of normal and disease contexts.
空间蛋白质组学有可能极大地促进我们对生物学、生理学和医学的理解。基质辅助激光解吸/电离质谱成像(MALDI-MSI)是空间蛋白质组学领域的强大工具,能够直接检测和记录组织中蛋白质的丰度和分布。MALDI-MSI 保留了空间分布和组织学,允许对复杂、异质的组织进行无偏分析。然而,MALDI-MSI 面临着同时进行肽定量和鉴定的挑战。为了克服这一挑战,我们开发并验证了 HIT-MAP(MALDI-MSI 蛋白质组学中的高分辨率信息学工具包),这是一个使用肽质量指纹分析和双重评分系统的开源生物信息学工作流程,用于计算将肽和蛋白质注释分配给高质量分辨率 MSI 数据集,并生成可定制的空间分布图谱。HIT-MAP 将成为空间蛋白质组学社区分析新生成和回顾性数据集的有价值资源,能够在广泛的正常和疾病背景下实现稳健的肽和蛋白质注释和可视化。