Petrosino Maria, Novak Leonore, Pasquo Alessandra, Chiaraluce Roberta, Turina Paola, Capriotti Emidio, Consalvi Valerio
Dipartimento Scienze Biochimiche "A. Rossi Fanelli", Sapienza University of Rome, 00185 Roma, Italy.
ENEA CR Frascati, Diagnostics and Metrology Laboratory FSN-TECFIS-DIM, 00044 Frascati, Italy.
Int J Mol Sci. 2021 May 21;22(11):5416. doi: 10.3390/ijms22115416.
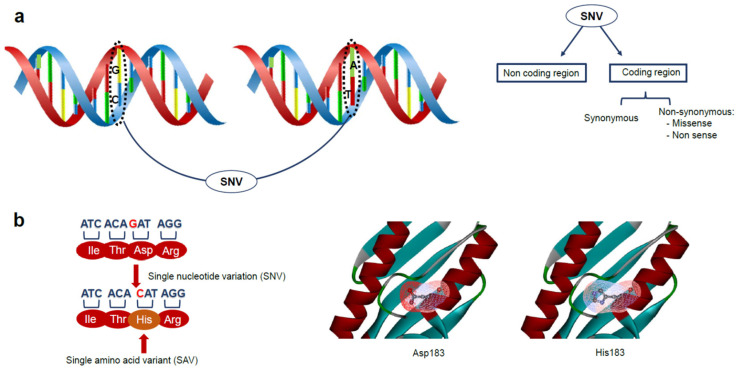
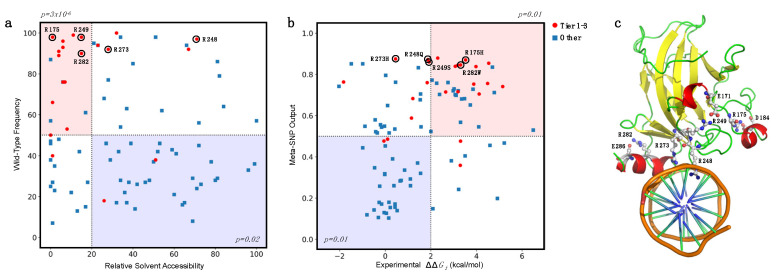
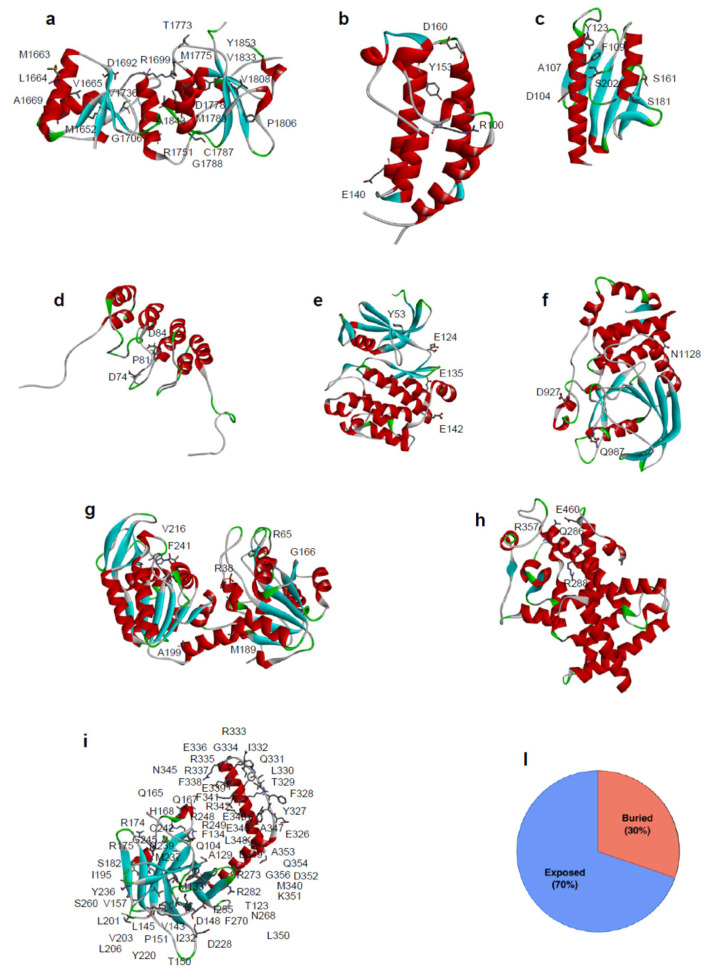
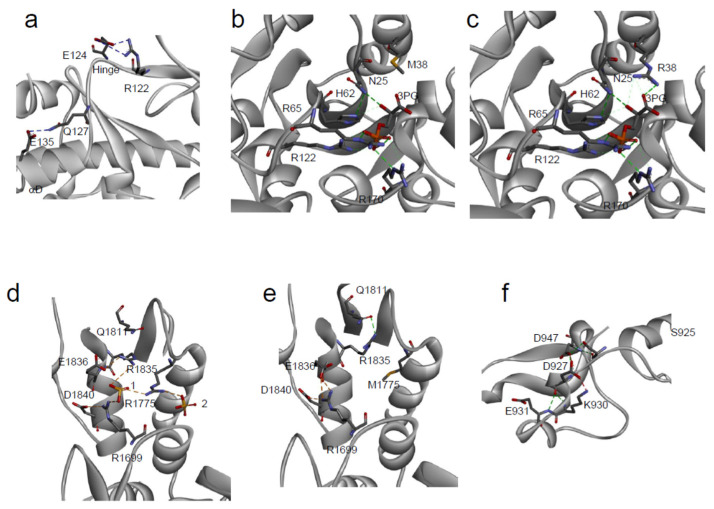
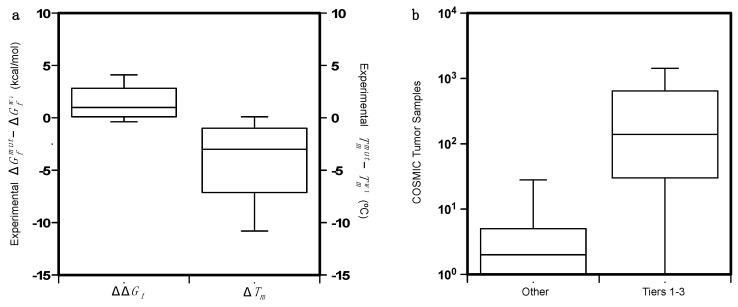
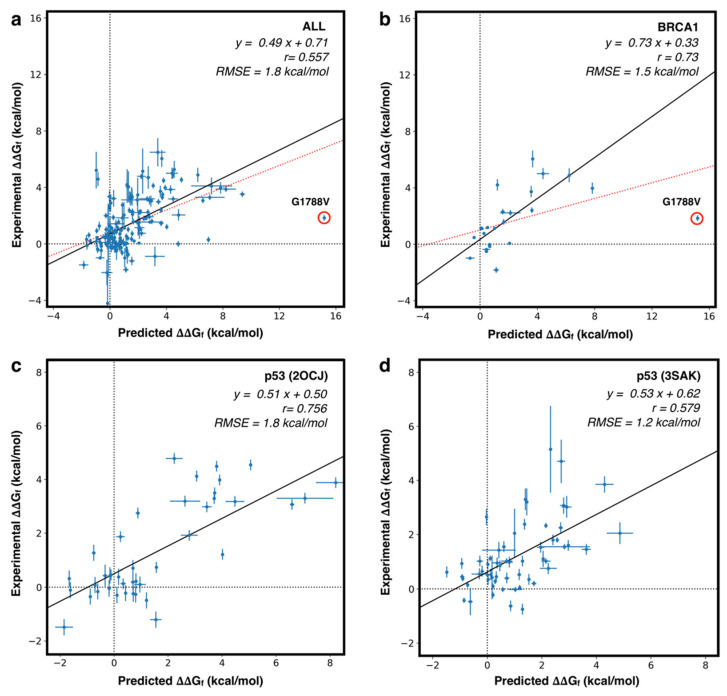
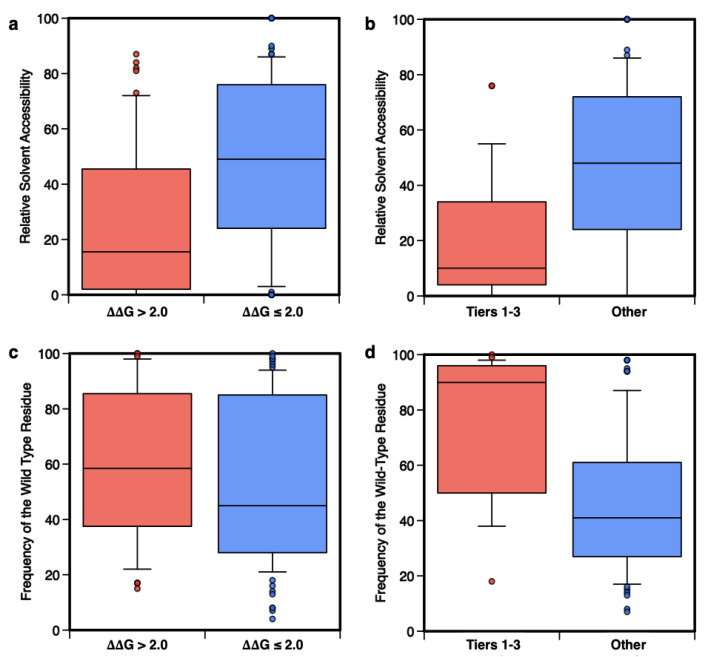
Large scale genome sequencing allowed the identification of a massive number of genetic variations, whose impact on human health is still unknown. In this review we analyze, by an in silico-based strategy, the impact of missense variants on cancer-related genes, whose effect on protein stability and function was experimentally determined. We collected a set of 164 variants from 11 proteins to analyze the impact of missense mutations at structural and functional levels, and to assess the performance of state-of-the-art methods (FoldX and Meta-SNP) for predicting protein stability change and pathogenicity. The result of our analysis shows that a combination of experimental data on protein stability and in silico pathogenicity predictions allowed the identification of a subset of variants with a high probability of having a deleterious phenotypic effect, as confirmed by the significant enrichment of the subset in variants annotated in the COSMIC database as putative cancer-driving variants. Our analysis suggests that the integration of experimental and computational approaches may contribute to evaluate the risk for complex disorders and develop more effective treatment strategies.
大规模基因组测序使得大量遗传变异得以识别,但其对人类健康的影响仍不明确。在本综述中,我们通过基于计算机模拟的策略,分析错义变异对癌症相关基因的影响,这些基因对蛋白质稳定性和功能的影响已通过实验确定。我们从11种蛋白质中收集了一组164个变异,以分析错义突变在结构和功能水平上的影响,并评估用于预测蛋白质稳定性变化和致病性的最新方法(FoldX和Meta-SNP)的性能。我们的分析结果表明,蛋白质稳定性实验数据与计算机模拟致病性预测相结合,能够识别出具有有害表型效应高可能性的变异子集,COSMIC数据库中注释为假定癌症驱动变异的变异子集中该子集的显著富集证实了这一点。我们的分析表明,实验方法与计算方法的整合可能有助于评估复杂疾病的风险,并制定更有效的治疗策略。