Department of Microbiology & Immunology, Schulich School of Medicine & Dentistry, University of Western Ontario, London, ON N6G 2V4, Canada.
Department of Biochemistry, Schulich School of Medicine & Dentistry, University of Western Ontario, London, ON N6G 2V4, Canada.
Cells. 2021 May 4;10(5):1101. doi: 10.3390/cells10051101.
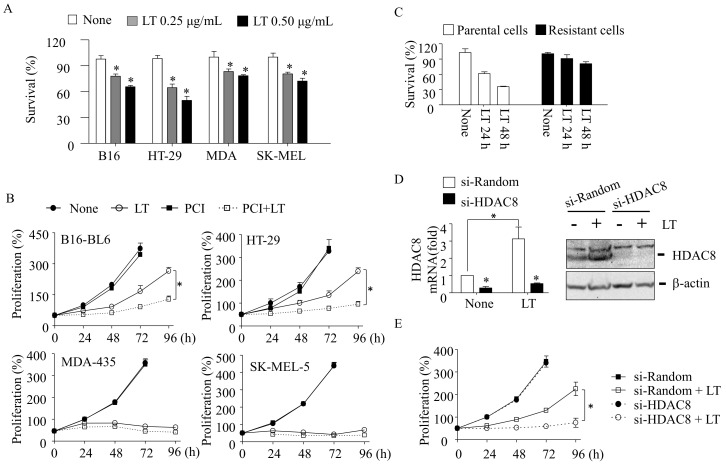
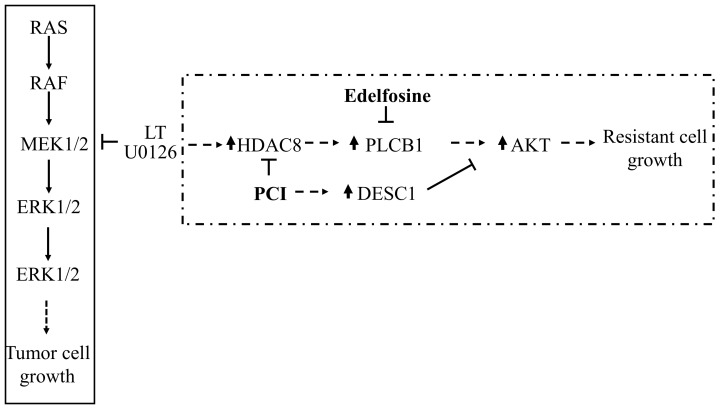
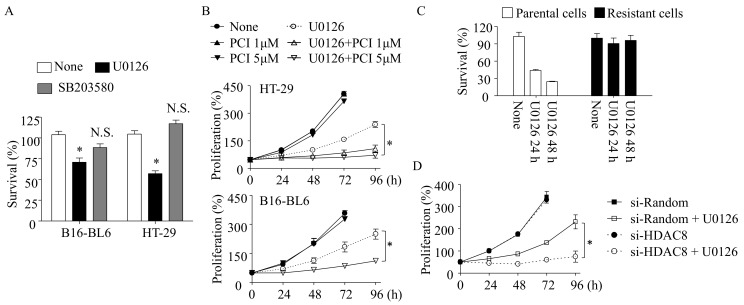
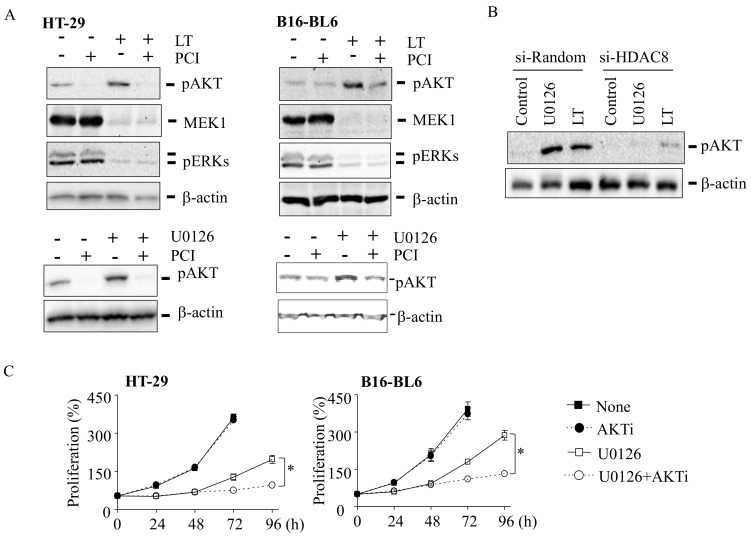
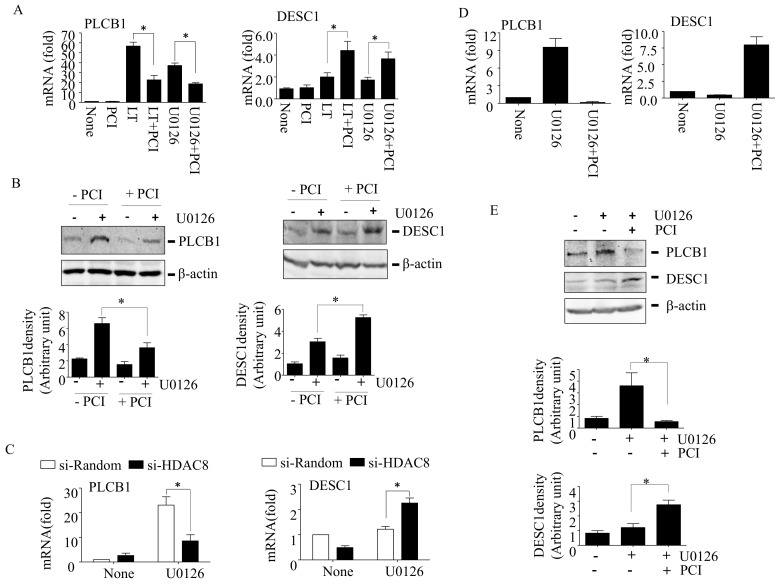
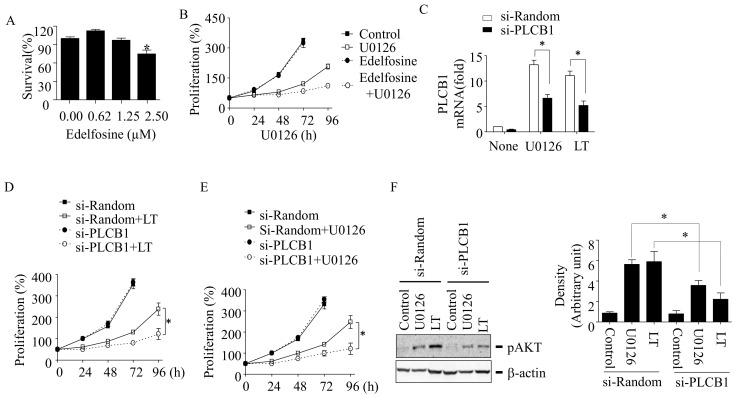
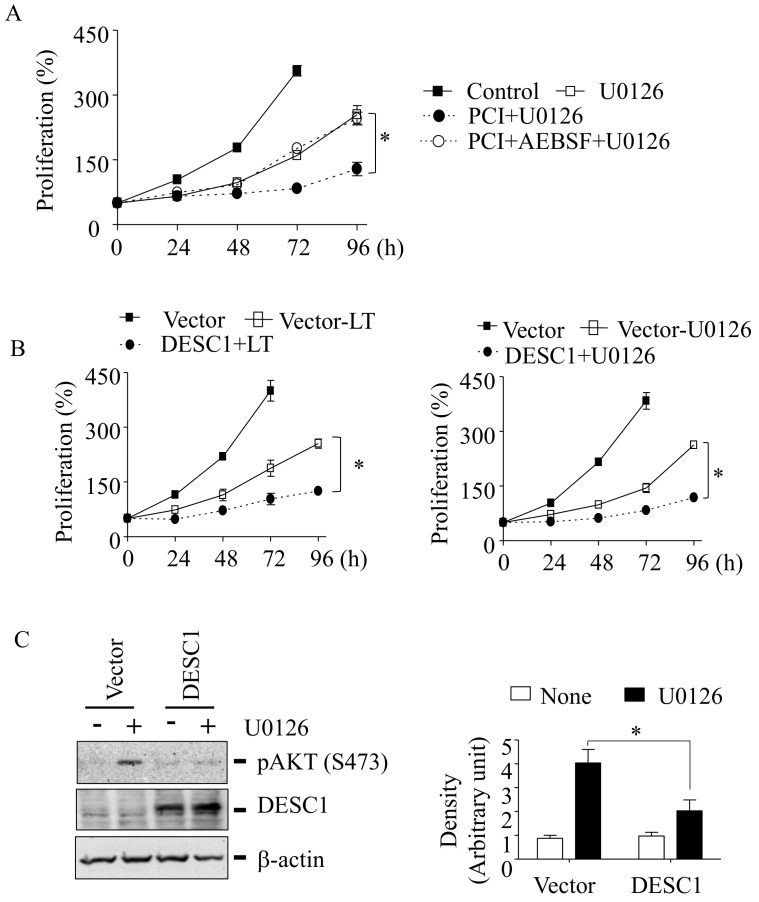
Inhibition of the RAF-MEK1/2-ERK signaling pathway is an ideal strategy for treating cancers with NRAS or BRAF mutations. However, the development of resistance due to incomplete inhibition of the pathway and activation of compensatory cell proliferation pathways is a major impediment of the targeted therapy. The anthrax lethal toxin (LT), which cleaves and inactivates MEKs, is a modifiable biomolecule that can be delivered selectively to tumor cells and potently kills various tumor cells. However, resistance to LT and the mechanism involved are yet to be explored. Here, we show that LT, through inhibiting MEK1/2-ERK activation, inhibits the proliferation of cancer cells with NRAS/BRAF mutations. Among them, the human colorectal tumor HT-29 and murine melanoma B16-BL6 cells developed resistance to LT in 2 to 3 days of treatment. These resistant cells activated AKT through a histone deacetylase (HDAC) 8-dependent pathway. Using an Affymetrix microarray, followed by qPCR validation, we identified that the differential expression of the phospholipase C-β1 (PLCB1) and squamous cell carcinoma-1 (DESC1) played an important role in HDAC8-mediated AKT activation and resistance to MEK1/2-ERK inhibition. By using inhibitors, small interference RNAs and/or expression vectors, we found that the inhibition of HDAC8 suppressed PLCB1 expression and induced DESC1 expression in the resistant cells, which led to the inhibition of AKT and re-sensitization to LT and MEK1/2 inhibition. These results suggest that targeting PLCB1 and DESC1 is a novel strategy for inhibiting the resistance to MEK1/2 inhibition.
抑制 RAF-MEK1/2-ERK 信号通路是治疗NRAS 或 BRAF 突变癌症的理想策略。然而,由于该通路不完全抑制和激活代偿性细胞增殖途径而导致的耐药性的发展是靶向治疗的主要障碍。炭疽致死毒素 (LT) 可切割并失活 MEKs,是一种可修饰的生物分子,可选择性递送至肿瘤细胞并有效地杀死各种肿瘤细胞。然而,LT 的耐药性及其涉及的机制仍有待探索。在这里,我们表明 LT 通过抑制 MEK1/2-ERK 的激活来抑制具有 NRAS/BRAF 突变的癌细胞的增殖。其中,人结直肠肿瘤 HT-29 和鼠黑色素瘤 B16-BL6 细胞在 2 至 3 天的治疗中对 LT 产生了耐药性。这些耐药细胞通过组蛋白去乙酰化酶 (HDAC)8 依赖性途径激活 AKT。通过使用 Affymetrix 微阵列,然后进行 qPCR 验证,我们确定了差异表达的磷酯酶 C-β1 (PLCB1) 和鳞状细胞癌-1 (DESC1) 在 HDAC8 介导的 AKT 激活和对 MEK1/2-ERK 抑制的耐药性中起着重要作用。通过使用抑制剂、小干扰 RNA 和/或表达载体,我们发现抑制 HDAC8 可抑制耐药细胞中 PLCB1 的表达并诱导 DESC1 的表达,从而抑制 AKT 并重新对 LT 和 MEK1/2 抑制敏感。这些结果表明,靶向 PLCB1 和 DESC1 是抑制 MEK1/2 抑制耐药性的一种新策略。