Department of Genetic and Molecular Medicine, Institut de Recerca, Hospital Sant Joan de Déu, 08950 Barcelona, Spain.
Laboratory of Molecular Physiology, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, 08003 Barcelona, Spain.
Int J Mol Sci. 2021 May 13;22(10):5180. doi: 10.3390/ijms22105180.
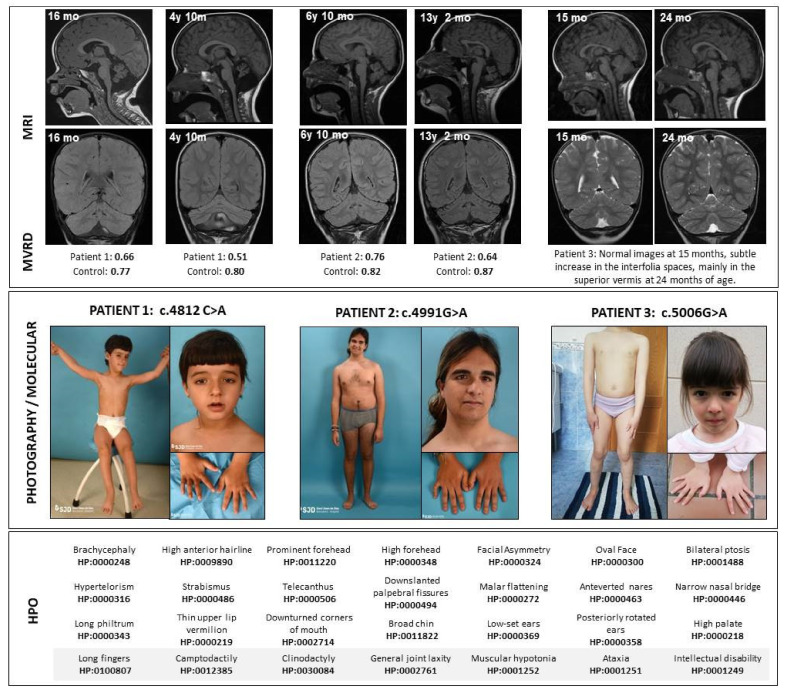
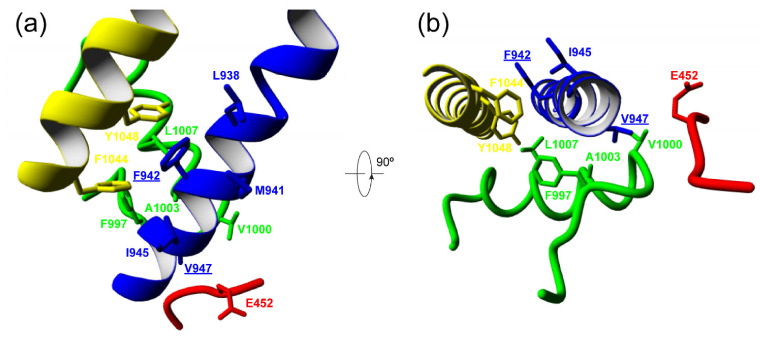
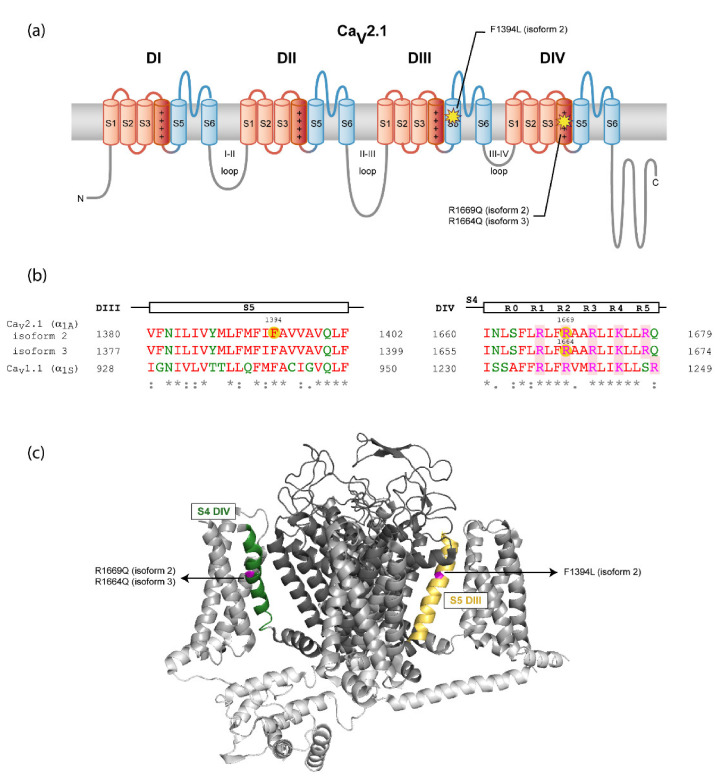
The gene encodes the pore-forming α subunit of the voltage-gated Ca2.1 Ca channel, essential in neurotransmission, especially in Purkinje cells. Mutations in result in great clinical heterogeneity with progressive symptoms, paroxysmal events or both. During infancy, clinical and neuroimaging findings may be unspecific, and no dysmorphic features have been reported. We present the clinical, radiological and evolutionary features of three patients with congenital ataxia, one of them carrying a new variant. We report the structural localization of variants and their expected functional consequences. There was an improvement in cerebellar syndrome over time despite a cerebellar atrophy progression, inconsistent response to acetazolamide and positive response to methylphenidate. The patients shared distinctive facial gestalt: oval face, prominent forehead, hypertelorism, downslanting palpebral fissures and narrow nasal bridge. The two α affected residues are fully conserved throughout evolution and among the whole human Ca channel family. They contribute to the channel pore and the voltage sensor segment. According to structural data analysis and available functional characterization, they are expected to exert gain- (F1394L) and loss-of-function (R1664Q/R1669Q) effect, respectively. Among the -related phenotypes, our results suggest that non-progressive congenital ataxia is associated with developmental delay and dysmorphic features, constituting a recognizable syndromic neurodevelopmental disorder.
该基因编码电压门控 Ca2.1 Ca 通道的孔形成 α 亚基,对神经传递至关重要,尤其是在浦肯野细胞中。的突变导致进行性症状、阵发性事件或两者兼有很大的临床异质性。在婴儿期,临床和神经影像学表现可能不特异,且尚未报道有发育异常特征。我们报告了 3 例先天性共济失调患者的临床、放射学和进化特征,其中 1 例携带新的变异。我们报告了变异的结构定位及其预期的功能后果。尽管小脑萎缩进展,但小脑综合征随时间推移得到改善,对乙酰唑胺的反应不一致,对哌醋甲酯的反应阳性。这些患者具有独特的面部特征:椭圆形脸、宽大的额头、远视、眼睑裂下斜和狭窄的鼻梁。受影响的两个 α 残基在整个进化过程中以及在整个人类 Ca 通道家族中都是完全保守的。它们有助于通道孔和电压传感器段。根据结构数据分析和可用的功能特征,预计它们分别发挥增益(F1394L)和功能丧失(R1664Q/R1669Q)效应。在 -相关表型中,我们的结果表明,非进行性先天性共济失调与发育迟缓、发育异常特征有关,构成一种可识别的综合征性神经发育障碍。