Quinn Taylor R, Steussy Calvin N, Haines Brandon E, Lei Jinping, Wang Wei, Sheong Fu Kit, Stauffacher Cynthia V, Huang Xuhui, Norrby Per-Ola, Helquist Paul, Wiest Olaf
Department of Chemistry and Biochemistry, University of Notre Dame Notre Dame IN 46556 USA
Early TDE Discovery, Early Oncology, Oncology R&D, AstraZeneca Boston USA.
Chem Sci. 2021 Apr 1;12(18):6413-6418. doi: 10.1039/d1sc00102g.
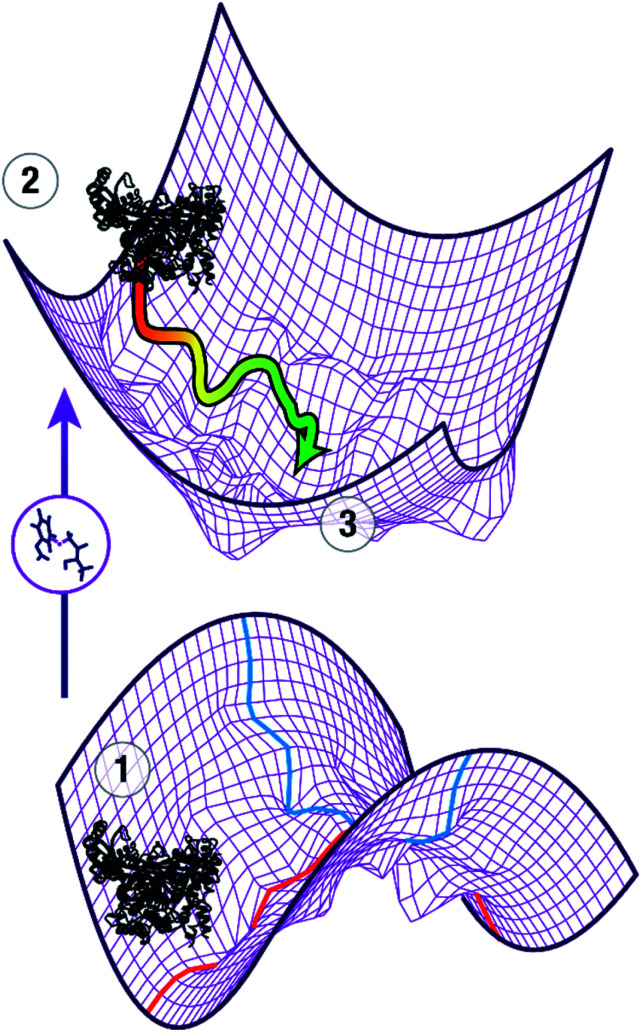
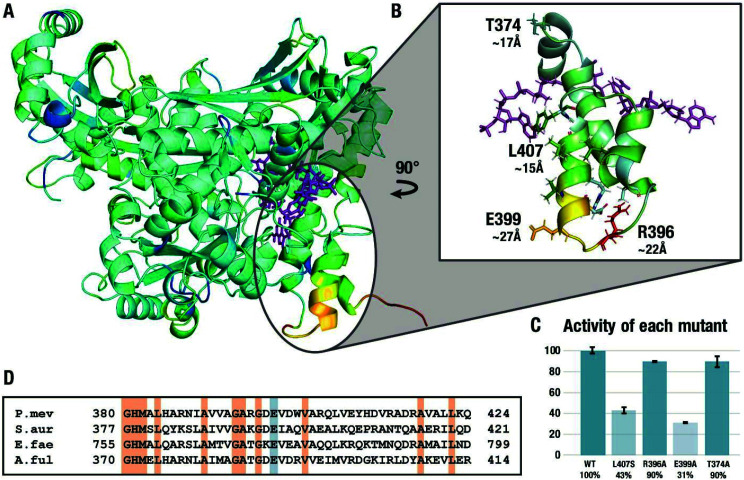
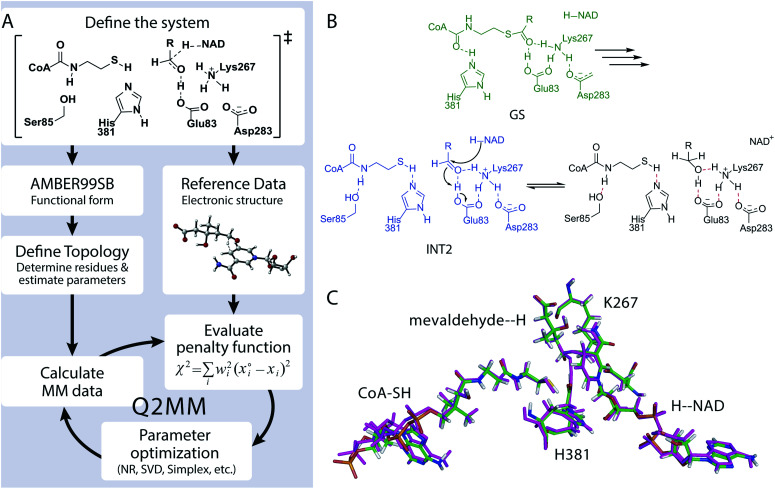
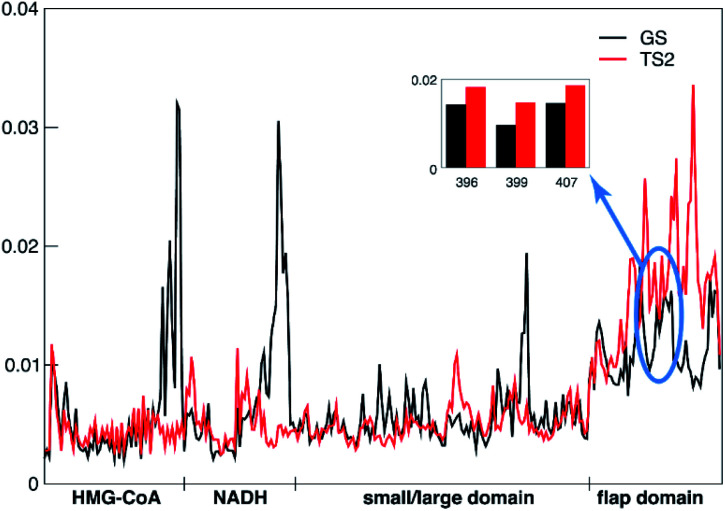
Understanding the mechanisms of enzymatic catalysis requires a detailed understanding of the complex interplay of structure and dynamics of large systems that is a challenge for both experimental and computational approaches. More importantly, the computational demands of QM/MM simulations mean that the dynamics of the reaction can only be considered on a timescale of nanoseconds even though the conformational changes needed to reach the catalytically active state happen on a much slower timescale. Here we demonstrate an alternative approach that uses transition state force fields (TSFFs) derived by the quantum-guided molecular mechanics (Q2MM) method that provides a consistent treatment of the entire system at the classical molecular mechanics level and allows simulations at the microsecond timescale. Application of this approach to the second hydride transfer transition state of HMG-CoA reductase from (HMGR) identified three remote residues, R396, E399 and L407, (15-27 Å away from the active site) that have a remote dynamic effect on enzyme activity. The predictions were subsequently validated experimentally site-directed mutagenesis. These results show that microsecond timescale MD simulations of transition states are possible and can predict rather than just rationalize remote allosteric residues.
理解酶催化机制需要详细了解大型系统结构与动力学之间的复杂相互作用,这对实验和计算方法而言都是一项挑战。更重要的是,量子力学/分子力学(QM/MM)模拟的计算需求意味着,即便达到催化活性状态所需的构象变化发生在慢得多的时间尺度上,反应动力学也只能在纳秒时间尺度上进行考虑。在此,我们展示了一种替代方法,该方法使用由量子引导分子力学(Q2MM)方法推导的过渡态力场(TSFFs),它能在经典分子力学水平上对整个系统进行一致处理,并允许在微秒时间尺度上进行模拟。将此方法应用于来自[具体来源未提及]的HMG - CoA还原酶(HMGR)的第二次氢化物转移过渡态,确定了三个远程残基,即R396、E399和L407(距离活性位点15 - 27 Å),它们对酶活性具有远程动态影响。随后通过定点诱变实验验证了这些预测。这些结果表明,过渡态的微秒时间尺度分子动力学模拟是可行的,并且能够预测而非仅仅解释远程变构残基。