Hsieh Hsin-Hui, Chen Yen-An, Chang Yao-Jen, Wang Hsin-Hui, Yu Ya-Han, Lin Sheng-Wei, Huang Yin-Jung, Lin Steven, Chang Ching-Jin
Graduate Institute of Biochemical Sciences, College of Life Science, National Taiwan University, No. 1 Sec 4 Roosevelt Rd, Taipei, 106, Taiwan.
Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan.
J Inflamm (Lond). 2021 Jun 5;18(1):22. doi: 10.1186/s12950-021-00288-2.
Tristetraprolin (TTP) family proteins contain conserved tandem CCCH zinc-finger binding to AU-rich elements and C-terminal NOT1-binding domain. TTP is phosphorylated extensively in cells, and its mRNA destabilization activity is regulated by protein phosphorylation.
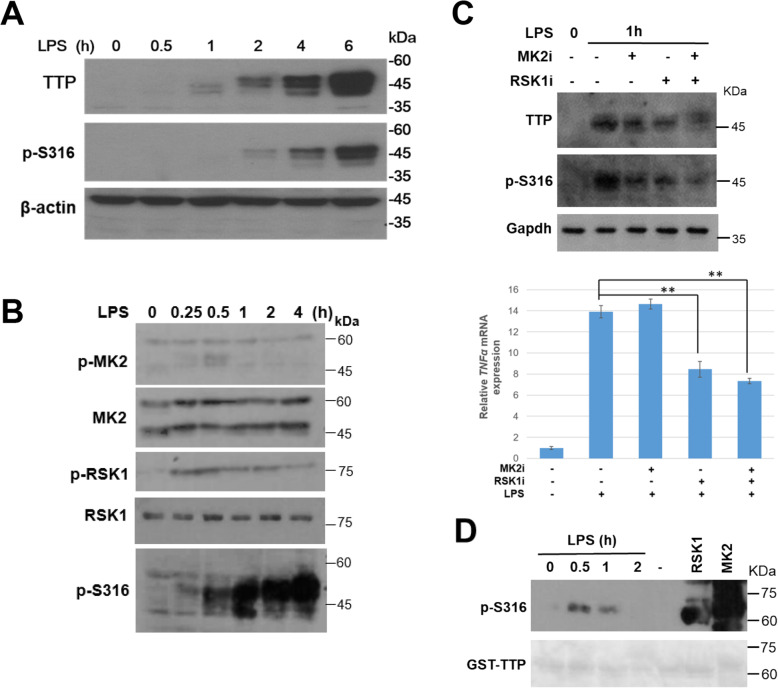
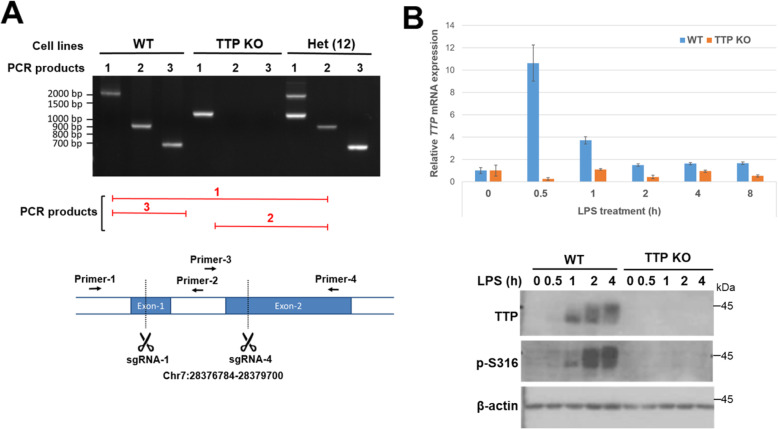
We generated an antibody against phospho-Serine316 located at the C-terminal NOT1-binding site and examined TTP phosphorylation in LPS-stimulated RAW264.7 cells. Knockout of TTP was created in RAW264.7 cells using CRISPR/Cas9 gene editing to explore TTP functions.
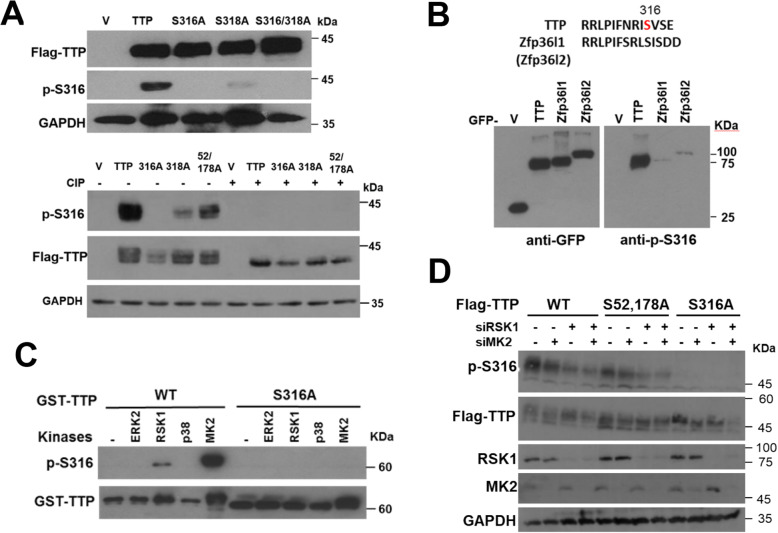
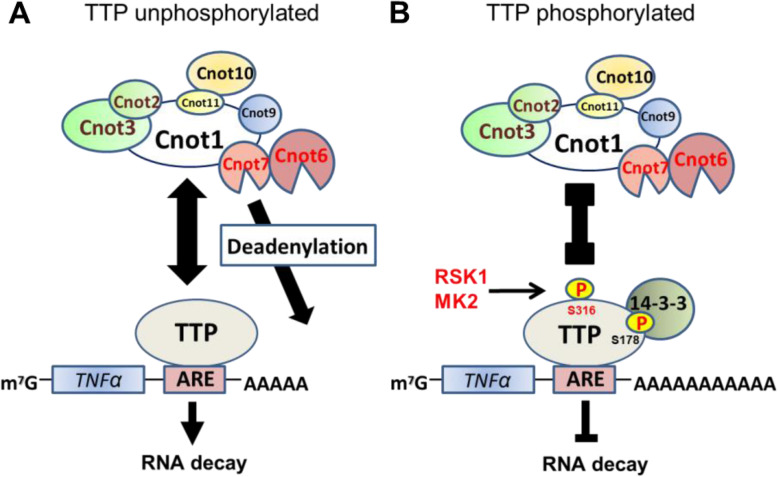
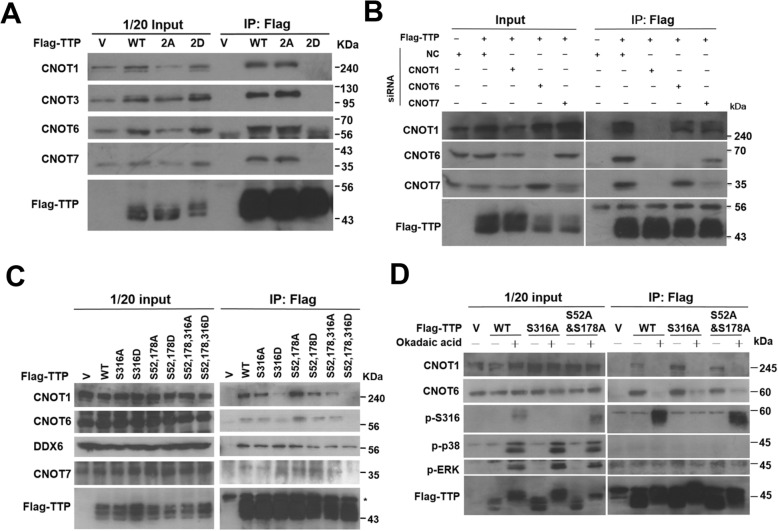
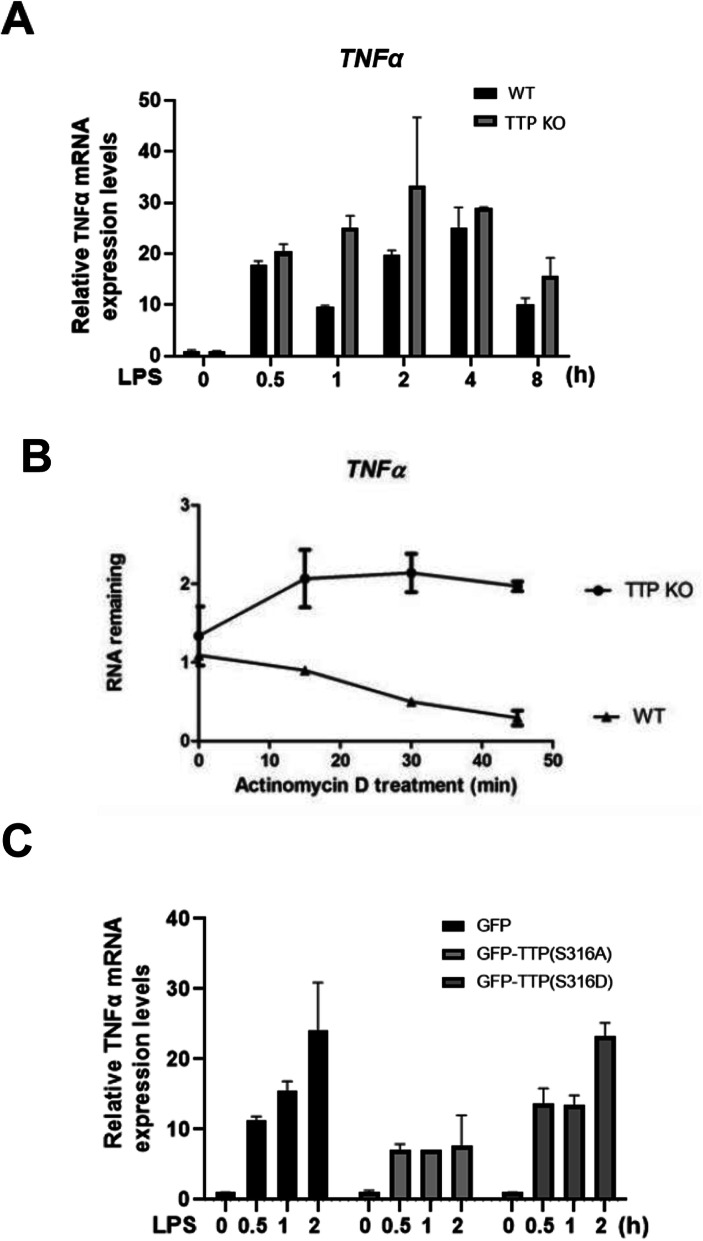
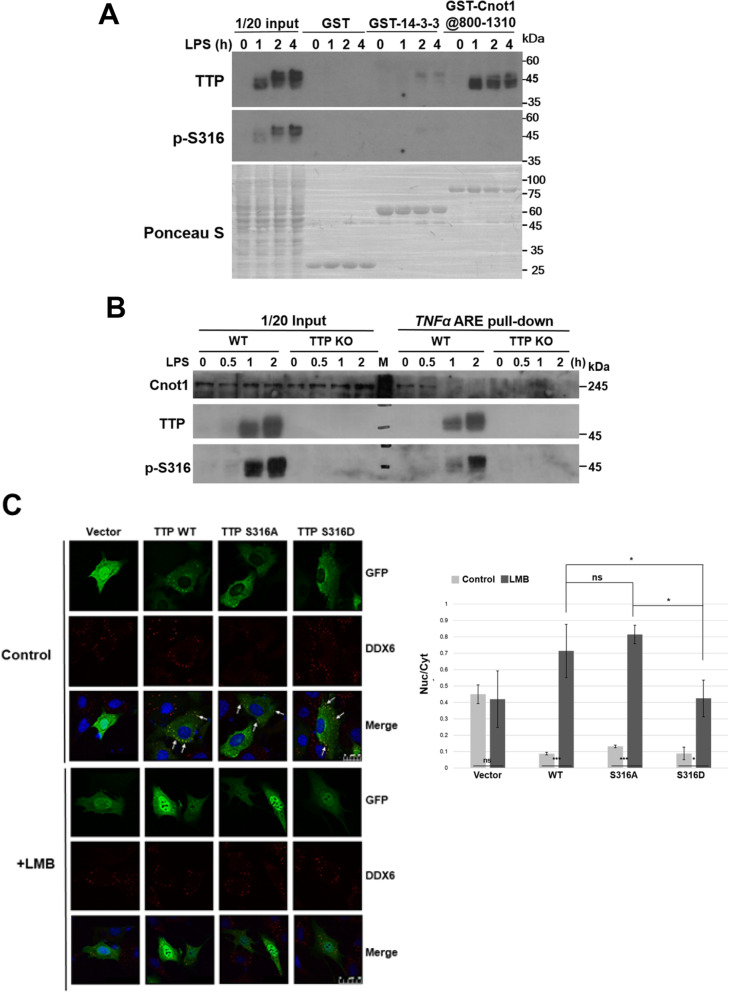
We demonstrated that Ser316 was phosphorylated by p90 ribosomal S6 kinase 1 (RSK1) and p38-activated protein kinase (MK2) and dephosphorylated by Protein Phosphatase 2A (PP2A). A phosphorylation-mimic mutant of S316D resulted in dissociation with the CCR4-NOT deadenylase complex through weakening interaction with CNOT1. Furthermore, Ser316 and serines 52 and 178 were independently contributed to the CCR4-NOT complex recruitment in the immunoprecipitation assay using phosphor-mimic mutants. In RAW264.7 macrophages, TTP was induced, and Ser316 was phosphorylated through RSK1 and MK2 by LPS stimulation. Knockout of TTP resulted in TNFα mRNA increased due to mRNA stabilization. Overexpression of non-phosphorylated S316A TTP mutant can restore TTP activity and lead to TNFα mRNA decreased. GST pull-down and RNA pull-down analyses demonstrated that endogenous TTP with Ser316 phosphorylation decreased the interaction with CNOT1.
Our results suggest that the TTP-mediated mRNA stability is modulated by Ser316 phosphorylation via regulating the TTP interaction with the CCR4-NOT deadenylase complex.
锌指蛋白(TTP)家族蛋白包含保守的串联CCCH锌指结构,可与富含AU的元件结合,以及C端NOT1结合结构域。TTP在细胞中被广泛磷酸化,其mRNA去稳定化活性受蛋白质磷酸化调控。
我们制备了一种针对位于C端NOT1结合位点的磷酸化丝氨酸316的抗体,并检测了脂多糖刺激的RAW264.7细胞中TTP的磷酸化情况。利用CRISPR/Cas9基因编辑技术在RAW264.7细胞中敲除TTP,以探究TTP的功能。
我们证明丝氨酸316被p90核糖体S6激酶1(RSK1)和p38激活的蛋白激酶(MK2)磷酸化,并被蛋白磷酸酶2A(PP2A)去磷酸化。S316D磷酸化模拟突变体通过减弱与CNOT1的相互作用,导致与CCR4-NOT去腺苷酸化酶复合物解离。此外,在使用磷酸化模拟突变体的免疫沉淀实验中,丝氨酸316以及丝氨酸52和178独立地促进了CCR4-NOT复合物的募集。在RAW264.7巨噬细胞中,TTP被诱导表达,并且脂多糖刺激通过RSK1和MK2使丝氨酸316磷酸化。敲除TTP导致由于mRNA稳定化,肿瘤坏死因子α(TNFα)mRNA增加。非磷酸化S316A TTP突变体的过表达可恢复TTP活性并导致TNFα mRNA减少。谷胱甘肽S-转移酶(GST)下拉和RNA下拉分析表明,丝氨酸316磷酸化的内源性TTP减少了与CNOT1的相互作用。
我们的结果表明,TTP介导的mRNA稳定性通过调节TTP与CCR4-NOT去腺苷酸化酶复合物的相互作用,受丝氨酸316磷酸化的调节。