Department of Epidemiology and Biostatistics, School of Public Health, Indiana University Bloomington, 1025 E. Seventh Street, Bloomington, 47405, IN, USA.
Hackensack-Meridian Health Center for Discovery and Innovation, Nutley, NJ, 07110, USA.
BMC Genom Data. 2021 Jun 10;22(1):20. doi: 10.1186/s12863-021-00975-2.
Most congenital heart defects (CHDs) result from complex interactions among genetic susceptibilities, epigenetic modifications, and maternal environmental exposures. Characterizing the complex relationship between genetic, epigenetic, and transcriptomic variation will enhance our understanding of pathogenesis in this important type of congenital disorder. We investigated cis-acting effects of genetic single nucleotide polymorphisms (SNPs) on local DNA methylation patterns within 83 cardiac tissue samples and prioritized their contributions to CHD risk by leveraging results of CHD genome-wide association studies (GWAS) and their effects on cardiac gene expression.
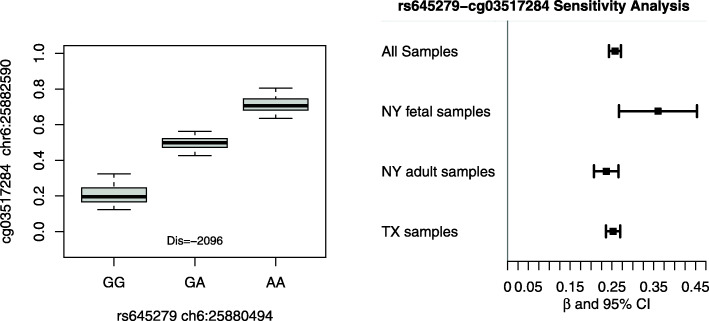
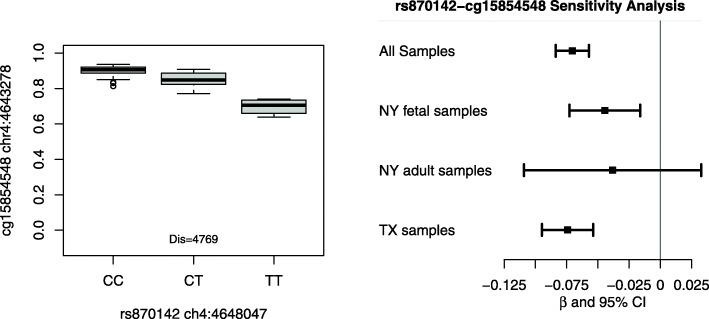
We identified 13,901 potential methylation quantitative trait loci (mQTLs) with a false discovery threshold of 5%. Further co-localization analyses and Mendelian randomization indicated that genetic variants near the HLA-DRB6 gene on chromosome 6 may contribute to CHD risk by regulating the methylation status of nearby CpG sites. Additional SNPs in genomic regions on chromosome 10 (TNKS2-AS1 gene) and chromosome 14 (LINC01629 gene) may simultaneously influence epigenetic and transcriptomic variations within cardiac tissues.
Our results support the hypothesis that genetic variants may influence the risk of CHDs through regulating the changes of DNA methylation and gene expression. Our results can serve as an important source of information that can be integrated with other genetic studies of heart diseases, especially CHDs.
大多数先天性心脏缺陷(CHD)是遗传易感性、表观遗传修饰和母体环境暴露之间复杂相互作用的结果。描述遗传、表观遗传和转录组变异之间的复杂关系将增强我们对这种重要类型先天性疾病发病机制的理解。我们研究了 83 个心脏组织样本中顺式作用的遗传单核苷酸多态性(SNP)对局部 DNA 甲基化模式的影响,并通过利用 CHD 全基因组关联研究(GWAS)的结果及其对心脏基因表达的影响,优先考虑它们对 CHD 风险的贡献。
我们鉴定了 13901 个具有 5%错误发现阈值的潜在甲基化数量性状基因座(mQTL)。进一步的共定位分析和孟德尔随机化表明,染色体 6 上 HLA-DRB6 基因附近的遗传变异可能通过调节附近 CpG 位点的甲基化状态来导致 CHD 风险。染色体 10(TNKS2-AS1 基因)和染色体 14(LINC01629 基因)上基因组区域的其他 SNP 可能同时影响心脏组织中的表观遗传和转录组变异。
我们的结果支持遗传变异可能通过调节 DNA 甲基化和基因表达的变化来影响 CHD 风险的假设。我们的结果可以作为与心脏病,特别是 CHD 的其他遗传研究相结合的重要信息来源。