Do Catherine, Shearer Alyssa, Suzuki Masako, Terry Mary Beth, Gelernter Joel, Greally John M, Tycko Benjamin
Institute for Cancer Genetics and Herbert Irving Comprehensive Cancer Center, Columbia University, New York, NY, 10032, USA.
Center for Epigenomics, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, 10461, USA.
Genome Biol. 2017 Jun 19;18(1):120. doi: 10.1186/s13059-017-1250-y.
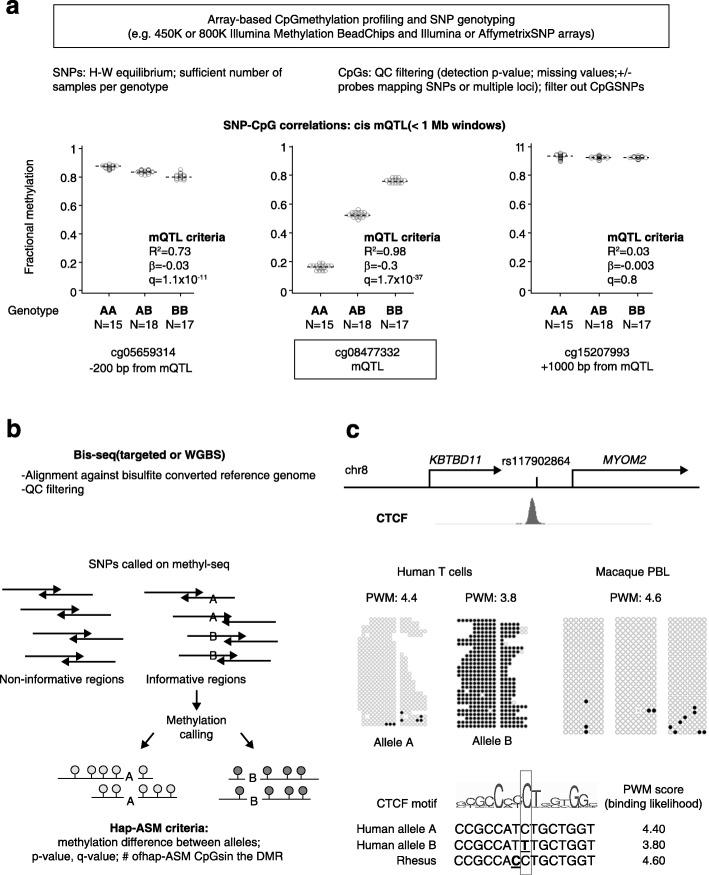
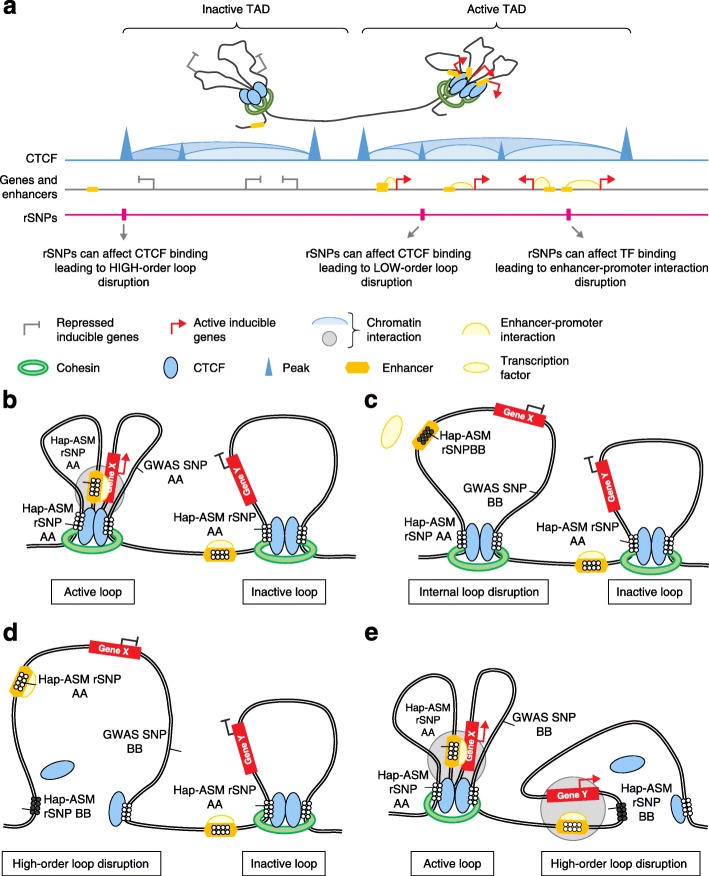
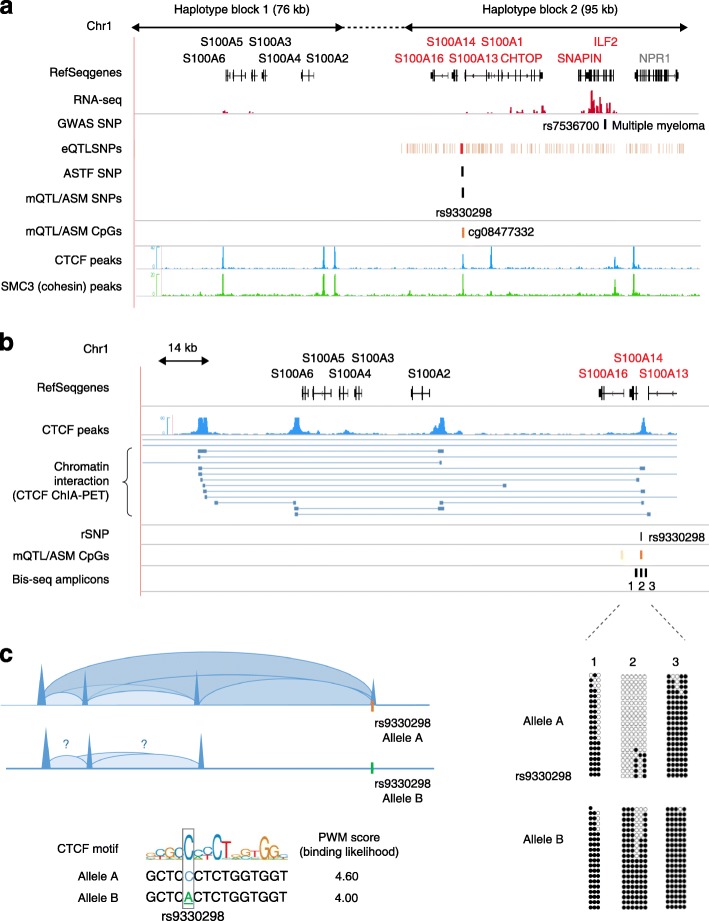
Studies on genetic-epigenetic interactions, including the mapping of methylation quantitative trait loci (mQTLs) and haplotype-dependent allele-specific DNA methylation (hap-ASM), have become a major focus in the post-genome-wide-association-study (GWAS) era. Such maps can nominate regulatory sequence variants that underlie GWAS signals for common diseases, ranging from neuropsychiatric disorders to cancers. Conversely, mQTLs need to be filtered out when searching for non-genetic effects in epigenome-wide association studies (EWAS). Sequence variants in CCCTC-binding factor (CTCF) and transcription factor binding sites have been mechanistically linked to mQTLs and hap-ASM. Identifying these sites can point to disease-associated transcriptional pathways, with implications for targeted treatment and prevention.
对基因-表观遗传相互作用的研究,包括甲基化数量性状位点(mQTL)图谱绘制和单倍型依赖的等位基因特异性DNA甲基化(hap-ASM),已成为全基因组关联研究(GWAS)后时代的主要焦点。这类图谱能够找出常见疾病GWAS信号背后的调控序列变异,这些疾病涵盖从神经精神疾病到癌症等多种类型。相反,在全表观基因组关联研究(EWAS)中寻找非遗传效应时,需要滤除mQTL。CCCTC结合因子(CTCF)和转录因子结合位点中的序列变异已在机制上与mQTL和hap-ASM相关联。识别这些位点能够指向与疾病相关的转录途径,对靶向治疗和预防具有重要意义。