Zhang Jinhui, Lu Haojie, Zhang Shuo, Wang Ting, Zhao Huashuo, Guan Fengjun, Zeng Ping
Department of Epidemiology and Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, China.
Center for Medical Statistics and Data Analysis, School of Public Health, Xuzhou Medical University, Xuzhou, China.
Front Genet. 2021 Jun 2;12:667877. doi: 10.3389/fgene.2021.667877. eCollection 2021.
Multiple genes were previously identified to be associated with cervical cancer; however, the genetic architecture of cervical cancer remains unknown and many potential causal genes are yet to be discovered.
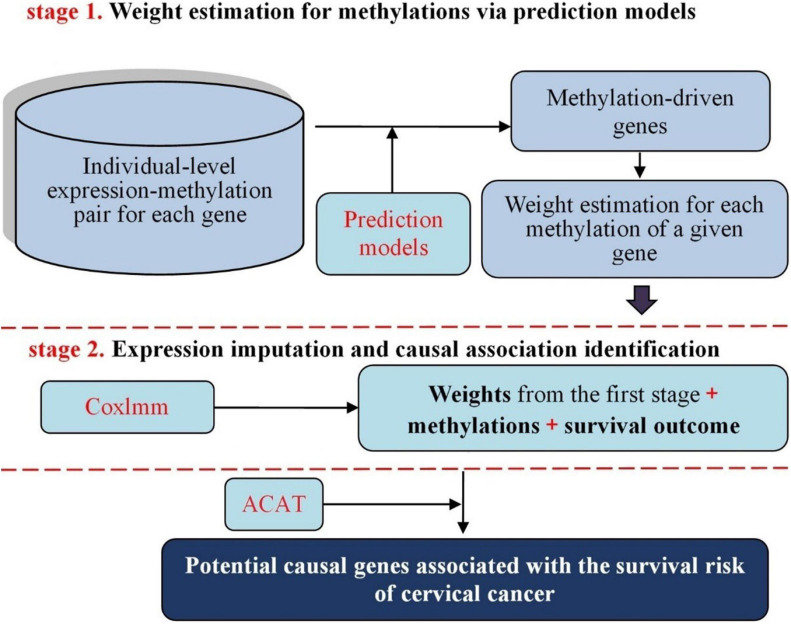
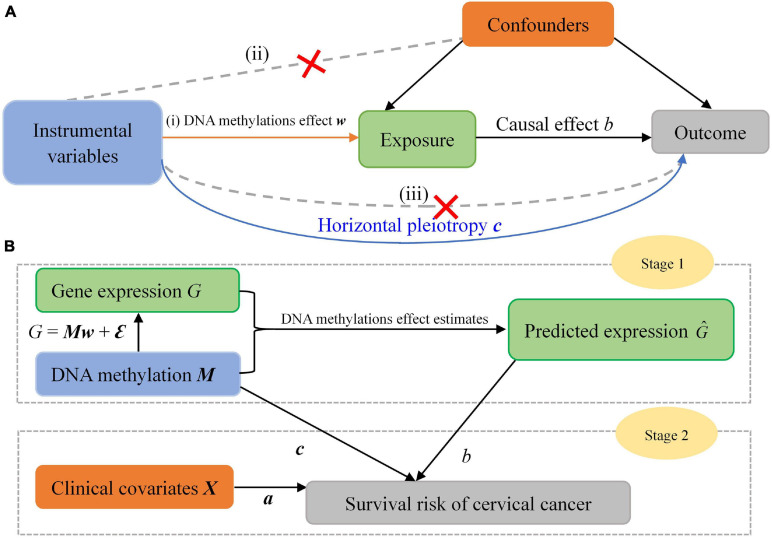
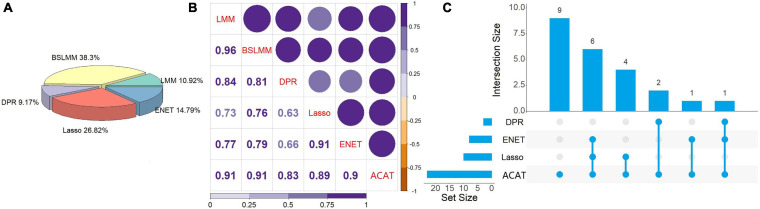
To explore potential causal genes related to cervical cancer, a two-stage causal inference approach was proposed within the framework of Mendelian randomization, where the gene expression was treated as exposure, with methylations located within the promoter regions of genes serving as instrumental variables. Five prediction models were first utilized to characterize the relationship between the expression and methylations for each gene; then, the methylation-regulated gene expression (MReX) was obtained and the association was evaluated via Cox mixed-effect model based on MReX. We further implemented the aggregated Cauchy association test (ACAT) combination to take advantage of respective strengths of these prediction models while accounting for dependency among the values.
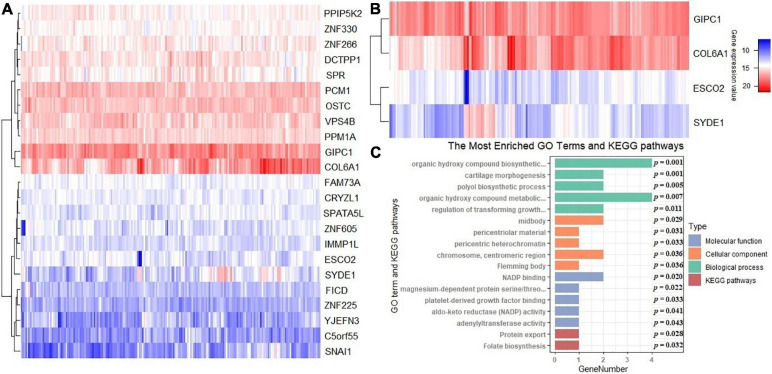
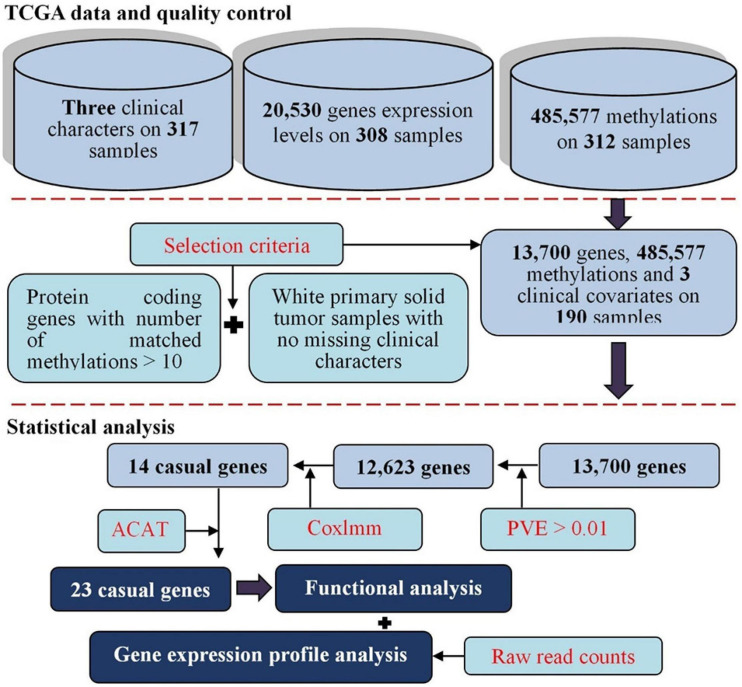
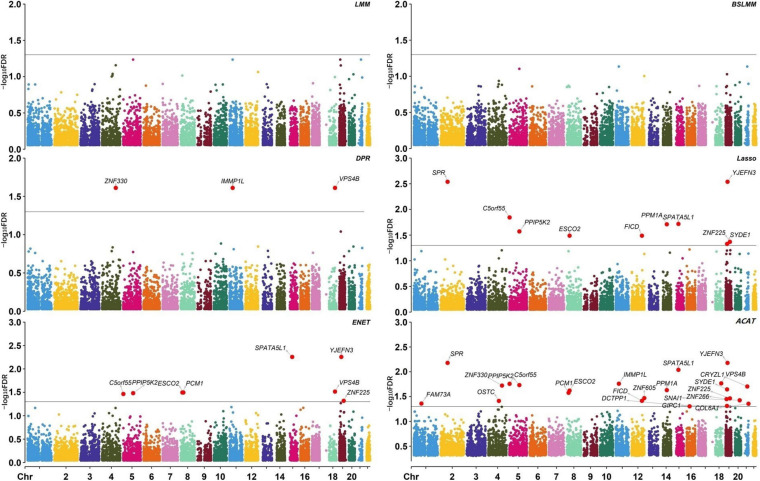
A total of 14 potential causal genes were discovered to be associated with the survival risk of cervical cancer in TCGA when the five prediction models were separately employed. The total number of potential causal genes was brought to 23 when conducting ACAT. Some of the newly discovered genes may be novel (e.g., , , , , , , , , , , , , , and ). Functional analyses showed that these genes were enriched in tumor-associated pathways. Additionally, four genes (i.e., , , , and ) were differentially expressed between tumor and normal tissues.
Our study discovered promising candidate genes that were causally associated with the survival risk of cervical cancer and thus provided new insights into the genetic etiology of cervical cancer.
先前已鉴定出多个与宫颈癌相关的基因;然而,宫颈癌的遗传结构仍不清楚,许多潜在的因果基因有待发现。
为了探索与宫颈癌相关的潜在因果基因,在孟德尔随机化框架内提出了一种两阶段因果推断方法,其中将基因表达视为暴露因素,将位于基因启动子区域内的甲基化作为工具变量。首先利用五个预测模型来表征每个基因的表达与甲基化之间的关系;然后,获得甲基化调节的基因表达(MReX),并基于MReX通过Cox混合效应模型评估关联。我们进一步实施了聚合柯西关联检验(ACAT)组合,以利用这些预测模型的各自优势,同时考虑值之间的依赖性。
当分别采用五个预测模型时,在TCGA中共发现14个潜在的因果基因与宫颈癌的生存风险相关。进行ACAT时,潜在因果基因的总数达到23个。一些新发现的基因可能是新的(例如, 、 、 、 、 、 、 、 、 、 、 、 、 和 )。功能分析表明,这些基因在肿瘤相关途径中富集。此外,四个基因(即 、 、 和 )在肿瘤组织和正常组织之间存在差异表达。
我们的研究发现了与宫颈癌生存风险有因果关系的有前景的候选基因,从而为宫颈癌的遗传病因提供了新的见解。