Kidney Center, Suzuka Kaisei Hospital, Suzuka, Japan.
Department of Cardiology and Nephrology, Mie University Graduate School of Medicine, 2-174 Edobashi, Tsu, Mie, 514-8507, Japan.
BMC Gastroenterol. 2021 Jun 24;21(1):267. doi: 10.1186/s12876-021-01845-y.
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent genetic kidney disease and polycystic liver disease is its major extrarenal manifestation, however biliary peritonitis due to a liver cyst rupture is extremely rare.
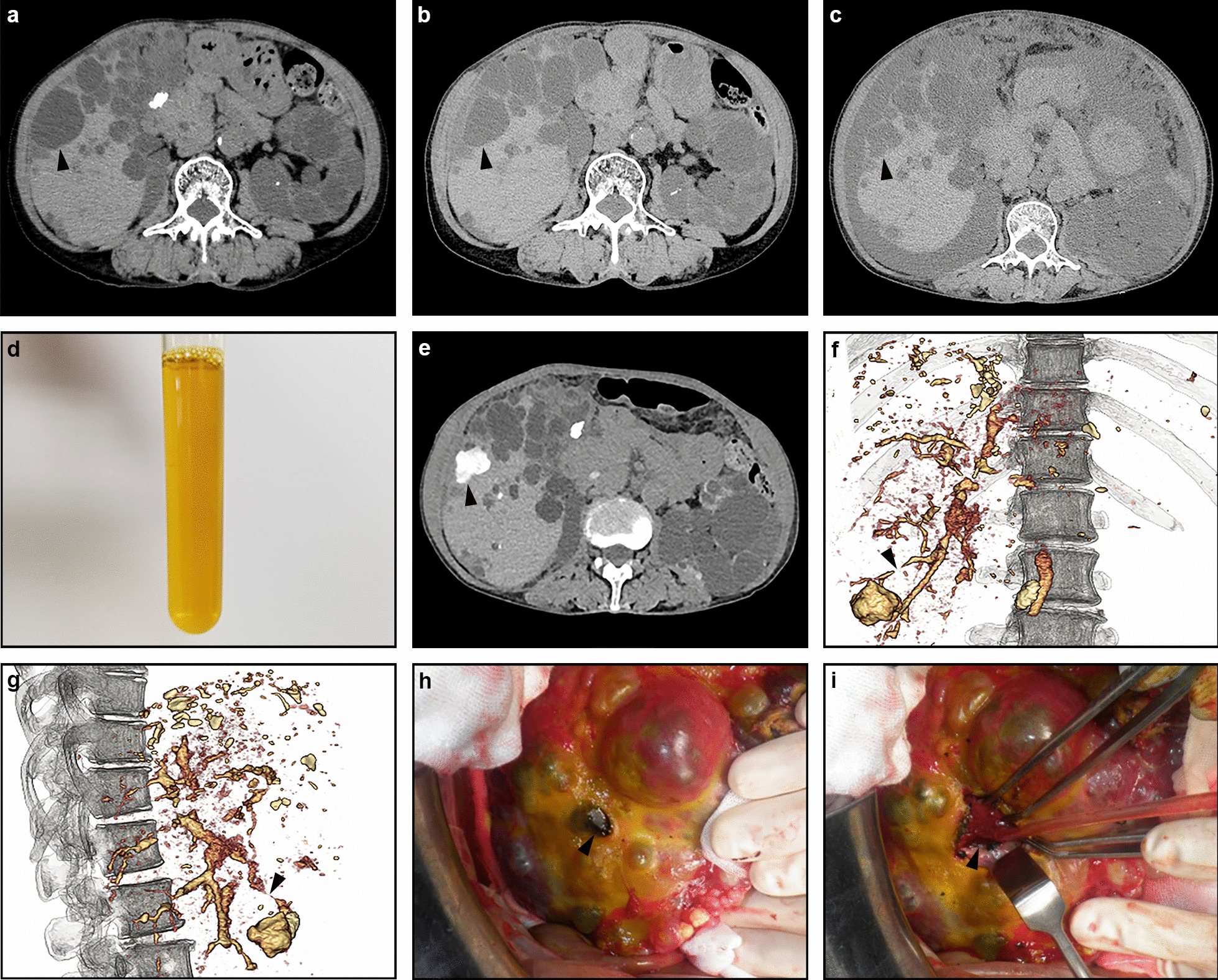
The patient was a 71-year-old Japanese woman who was diagnosed with ADPKD 3 years previously and developed right abdominal pain suddenly 1 month previously. As abdominal computed tomography (CT) showed a ruptured liver cyst in the right lobe, she was admitted to our hospital. Her symptoms improved with conservative management and she was discharged from the hospital after 1 week. Although she was asymptomatic for a while, she noticed abdominal distension and general malaise at 1 month after hospital discharge. Since abdominal CT showed massive ascites, she was admitted to our hospital again. A physical examination revealed abdominal distention without tenderness. Her serum creatinine, alkaline phosphatase, γ-glutamyl transpeptidase, total bilirubin, and CA19-9 were elevated. Abdominal paracentesis revealed amber transparent ascites and the bilirubin and CA19-9 concentrations were high. She was diagnosed with biliary peritonitis due to a ruptured liver cyst. Hemodialysis treatment was initiated with drainage of the ascites. The outflow of the ascites was no tendency to decrease and drip infusion cholangiography (DIC)-CT revealed a communication between the ruptured cyst and an intrahepatic bile duct. On day 31, she was transferred to a university hospital and abdominal surgery was performed. After removing the necrotic roof of the ruptured cyst on the right liver lobe, the orifice of the bile leakage was sutured. Cholecystectomy was performed and cholangiography showed no stones in the common bile duct. Abdominal CT one month after the operation showed no recurrence of ascites and she was discharged on day 49. Hemodialysis treatment was discontinued immediately after discharge because urine volume increased and her creatinine level decreased. There has been no recurrence of ascites since then.
While rare, biliary peritonitis can occur in association with the rupture of a liver cyst in ADPKD patients due to communication between the cyst and the intrahepatic bile duct, and DIC-CT should be recommended when biliary cyst rupture is suspected.
常染色体显性多囊肾病(ADPKD)是最常见的遗传性肾脏疾病,多囊性肝病是其主要的肾外表现,然而,由于肝囊肿破裂导致的胆汁性腹膜炎极为罕见。
患者为一名 71 岁的日本女性,3 年前被诊断为 ADPKD,并在 1 个月前突然出现右腹痛。由于腹部计算机断层扫描(CT)显示右叶肝囊肿破裂,她被收入我院。经保守治疗后,她的症状得到改善,并在 1 周后出院。尽管她在一段时间内无症状,但在出院后 1 个月时她注意到腹胀和全身不适。由于腹部 CT 显示大量腹水,她再次被收入我院。体格检查显示腹胀,无压痛。她的血清肌酐、碱性磷酸酶、γ-谷氨酰转肽酶、总胆红素和 CA19-9 升高。腹腔穿刺抽出琥珀色透明腹水,胆红素和 CA19-9 浓度较高。她被诊断为由于肝囊肿破裂导致的胆汁性腹膜炎。开始进行血液透析治疗以引流腹水。腹水流出没有减少的趋势,经胆管滴注造影(DIC)-CT 显示破裂囊肿与肝内胆管之间存在相通。在第 31 天,她被转至一所大学医院并进行了腹部手术。在切除右肝叶破裂囊肿的坏死顶部后,缝合了胆汁漏出的口。进行了胆囊切除术,胆管造影显示胆总管内无结石。术后 1 个月的腹部 CT 显示无腹水复发,她在第 49 天出院。出院后立即停止血液透析治疗,因为尿量增加,肌酐水平下降。此后,腹水未再复发。
虽然罕见,但由于囊肿与肝内胆管相通,ADPKD 患者的肝囊肿破裂可导致胆汁性腹膜炎,当怀疑胆管囊肿破裂时,应推荐进行 DIC-CT。