Interdisciplinary Program in Statistics and Data Science, University of Arizona, 617 N. Santa Rita Ave., 85721, Tucson, USA.
Department of Mathematics, University of Arizona, 617 N. Santa Rita Ave., 85721, Tucson, USA.
BMC Bioinformatics. 2021 Jun 26;22(1):348. doi: 10.1186/s12859-021-04265-7.
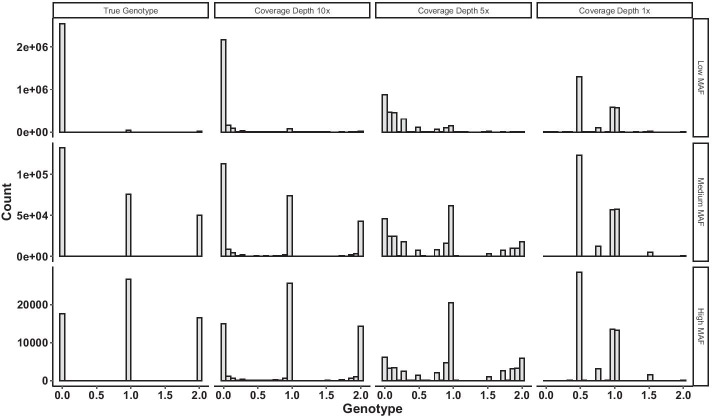
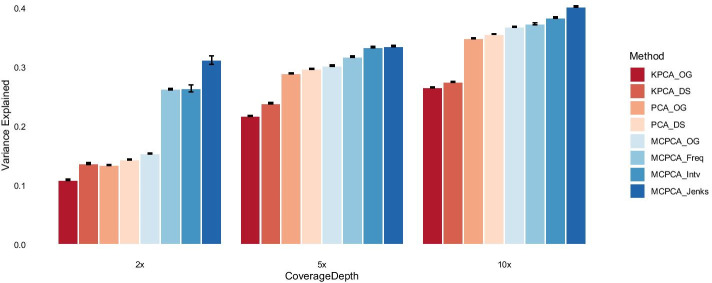
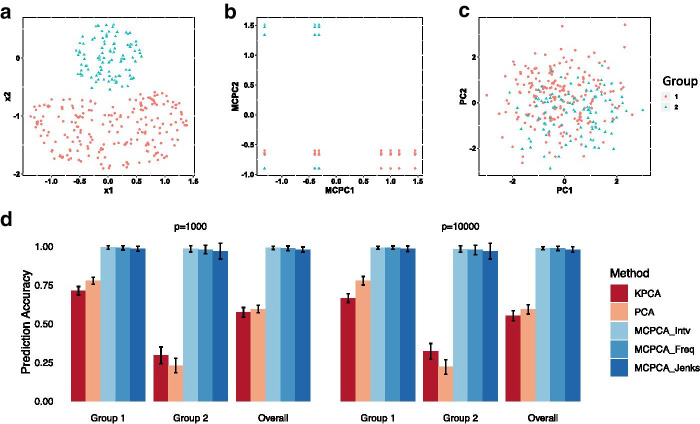
Low-depth sequencing allows researchers to increase sample size at the expense of lower accuracy. To incorporate uncertainties while maintaining statistical power, we introduce MCPCA_PopGen to analyze population structure of low-depth sequencing data.
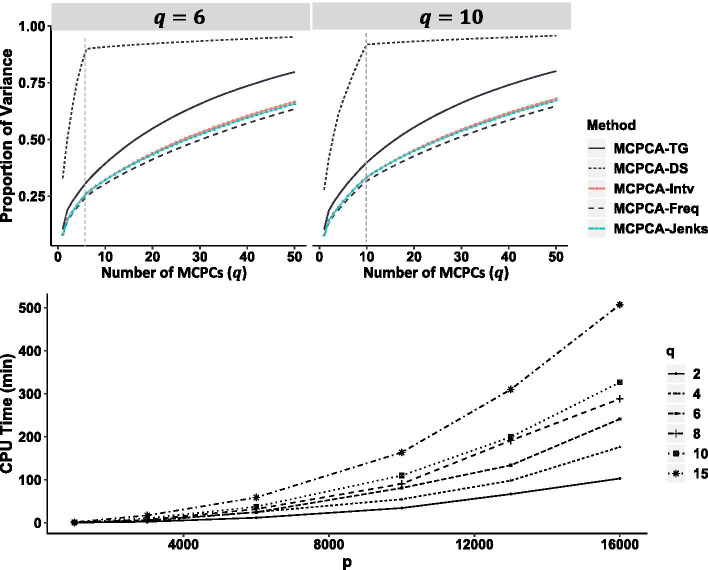
The method optimizes the choice of nonlinear transformations of dosages to maximize the Ky Fan norm of the covariance matrix. The transformation incorporates the uncertainty in calling between heterozygotes and the common homozygotes for loci having a rare allele and is more linear when both variants are common.
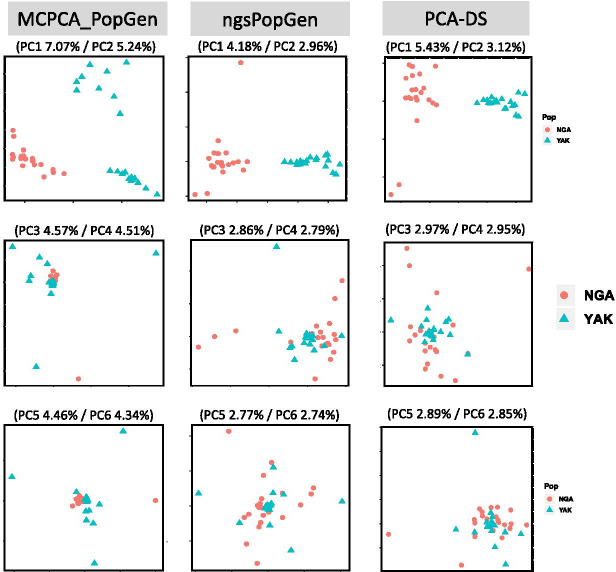
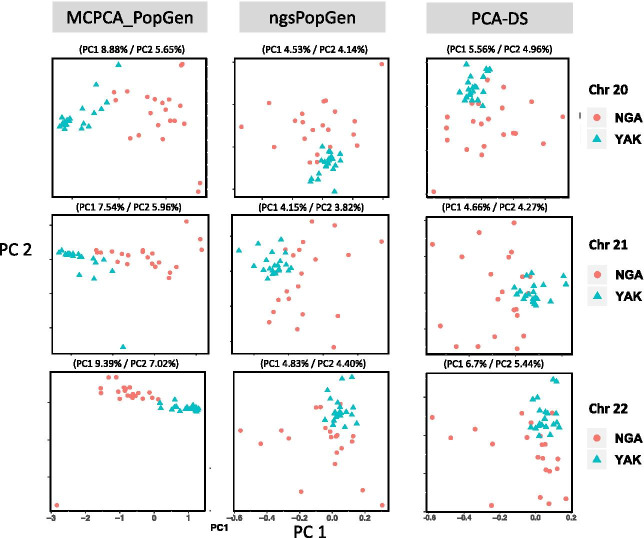
We apply MCPCA_PopGen to samples from two indigenous Siberian populations and reveal hidden population structure accurately using only a single chromosome. The MCPCA_PopGen package is available on https://github.com/yiwenstat/MCPCA_PopGen .
低深度测序允许研究人员以牺牲准确性为代价来增加样本量。为了在保持统计功效的同时纳入不确定性,我们引入了 MCPCA_PopGen 来分析低深度测序数据的群体结构。
该方法优化了剂量的非线性变换选择,以最大化协方差矩阵的 Ky Fan 范数。这种变换结合了稀有等位基因位点中杂合子和常见纯合子之间的调用不确定性,并且在两种变体都很常见时更加线性。
我们将 MCPCA_PopGen 应用于来自两个西伯利亚原住民群体的样本,并仅使用单个染色体准确地揭示隐藏的群体结构。MCPCA_PopGen 软件包可在 https://github.com/yiwenstat/MCPCA_PopGen 上获得。