Animal Breeding and Genomics, Wageningen University & Research, P.O. Box 338, 6700 AH, Wageningen, The Netherlands.
Genet Sel Evol. 2018 Jun 26;50(1):34. doi: 10.1186/s12711-018-0404-z.
High levels of pairwise linkage disequilibrium (LD) in single nucleotide polymorphism (SNP) array or whole-genome sequence data may affect both performance and efficiency of genomic prediction models. Thus, this warrants pruning of genotyping data for high LD. We developed an algorithm, named SNPrune, which enables the rapid detection of any pair of SNPs in complete or high LD throughout the genome.
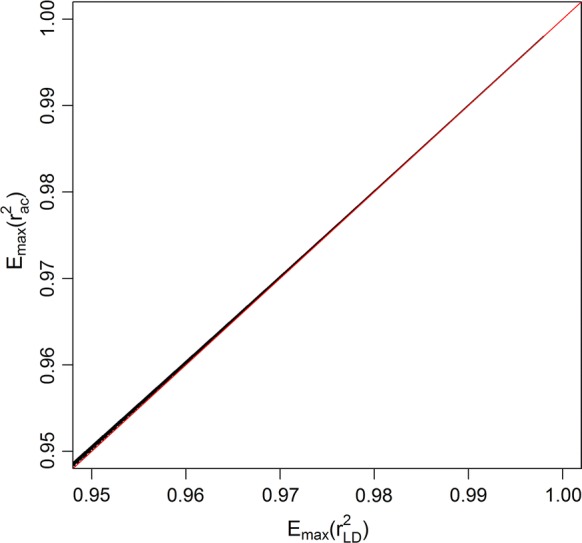
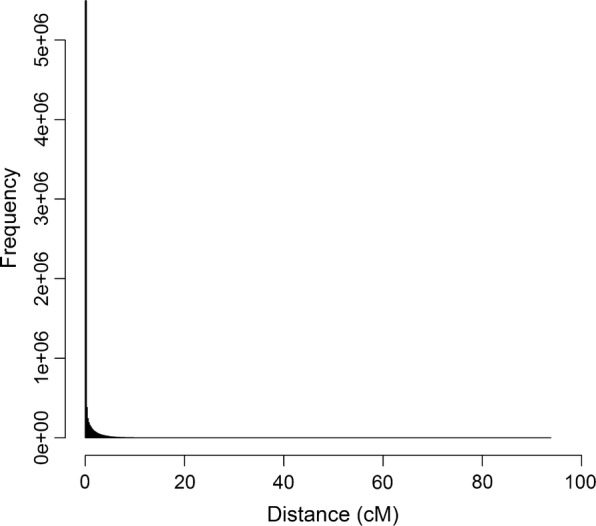
LD, measured as the squared correlation between phased alleles (r), can only reach a value of 1 when both loci have the same count of the minor allele. Sorting loci based on the minor allele count, followed by comparison of their alleles, enables rapid detection of loci in complete LD. Detection of loci in high LD can be optimized by computing the range of the minor allele count at another locus for each possible value of the minor allele count that can yield LD values higher than a predefined threshold. This efficiently reduces the number of pairs of loci for which LD needs to be computed, instead of considering all pairwise combinations of loci. The implemented algorithm SNPrune considered bi-allelic loci either using phased alleles or allele counts as input. SNPrune was validated against PLINK on two datasets, using an r threshold of 0.99. The first dataset contained 52k SNP genotypes on 3534 pigs and the second dataset contained simulated whole-genome sequence data with 10.8 million SNPs and 2500 animals.
SNPrune removed a similar number of SNPs as PLINK from the pig data but SNPrune was almost 12 times faster than PLINK. From the simulated sequence data with 10.8 million SNPs, SNPrune removed 6.4 and 1.4 million SNPs due to complete and high LD. Results were very similar regardless of whether phased alleles or allele counts were used. Using allele counts and multi-threading with 10 threads, SNPrune completed the analysis in 21 min. Using a sliding window of up to 500,000 SNPs, PLINK removed ~ 43,000 less SNPs (0.6%) in the sequence data and SNPrune was 24 to 170 times faster, using one or ten threads, respectively.
The SNPrune algorithm developed here is able to remove SNPs in high LD throughout the genome very efficiently in large datasets.
在单核苷酸多态性 (SNP) 数组或全基因组序列数据中,高水平的成对连锁不平衡 (LD) 可能会影响基因组预测模型的性能和效率。因此,有必要对基因型数据进行高 LD 修剪。我们开发了一种名为 SNPrune 的算法,该算法可以快速检测整个基因组中任何一对处于完全或高度 LD 的 SNP。
LD 是指相位等位基因之间的平方相关系数 (r),只有当两个基因座具有相同数量的次要等位基因时,LD 才能达到 1 的值。基于次要等位基因数量对基因座进行排序,然后比较它们的等位基因,可以快速检测完全 LD 的基因座。通过计算另一个基因座的次要等位基因计数的范围,对于每个可能产生高于预定义阈值的 LD 值的次要等位基因计数,可以优化对高度 LD 基因座的检测。这有效地减少了需要计算 LD 的基因座对的数量,而不是考虑基因座的所有成对组合。实现的算法 SNPrune 考虑了使用相位等位基因或等位基因计数作为输入的双等位基因基因座。我们使用 r 阈值为 0.99 在两个数据集上对 PLINK 进行了验证。第一个数据集包含 3534 头猪的 52k SNP 基因型,第二个数据集包含模拟的全基因组序列数据,其中包含 1080 万个 SNPs 和 2500 头动物。
SNPrune 从猪数据中删除了与 PLINK 相似数量的 SNP,但 SNPrune 的速度几乎比 PLINK 快 12 倍。对于包含 1080 万个 SNPs 的模拟序列数据,由于完全和高度 LD,SNPrune 删除了 640 万和 140 万个 SNP。无论使用相位等位基因还是等位基因计数,结果都非常相似。使用等位基因计数和 10 个线程的多线程,SNPrune 在 21 分钟内完成了分析。使用多达 50 万个 SNPs 的滑动窗口,PLINK 在序列数据中删除了约 43000 个 (0.6%)较少的 SNP,而 SNPrune 使用一个或十个线程的速度分别快 24 到 170 倍。
本文开发的 SNPrune 算法能够在大型数据集高效地去除基因组中处于高度 LD 的 SNP。