Frisby Trevor S, Langmead Christopher James
Computational Biology Department, School of Computer Science, Carnegie Mellon University, 5000 Forbes Ave, Pittsburgh, PA, 15213, USA.
Algorithms Mol Biol. 2021 Jul 1;16(1):13. doi: 10.1186/s13015-021-00195-4.
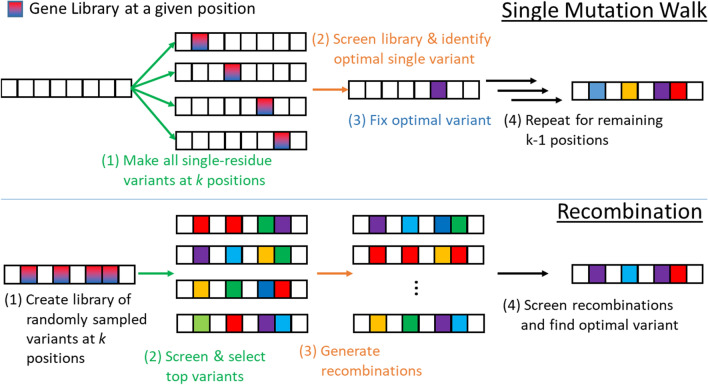
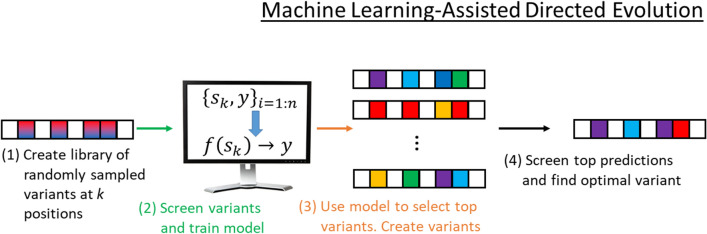
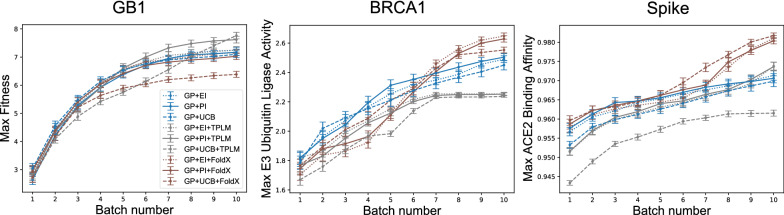
Directed evolution (DE) is a technique for protein engineering that involves iterative rounds of mutagenesis and screening to search for sequences that optimize a given property, such as binding affinity to a specified target. Unfortunately, the underlying optimization problem is under-determined, and so mutations introduced to improve the specified property may come at the expense of unmeasured, but nevertheless important properties (ex. solubility, thermostability, etc). We address this issue by formulating DE as a regularized Bayesian optimization problem where the regularization term reflects evolutionary or structure-based constraints.
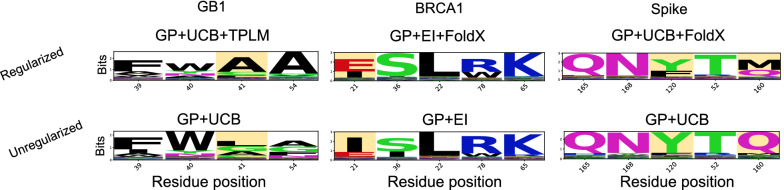
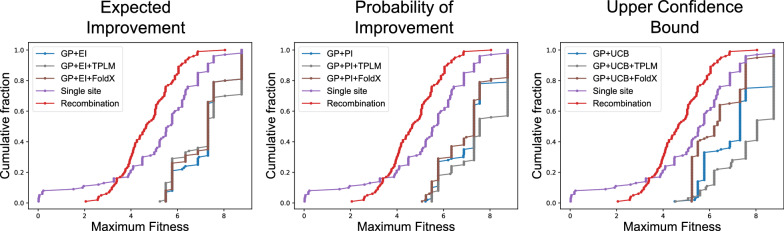
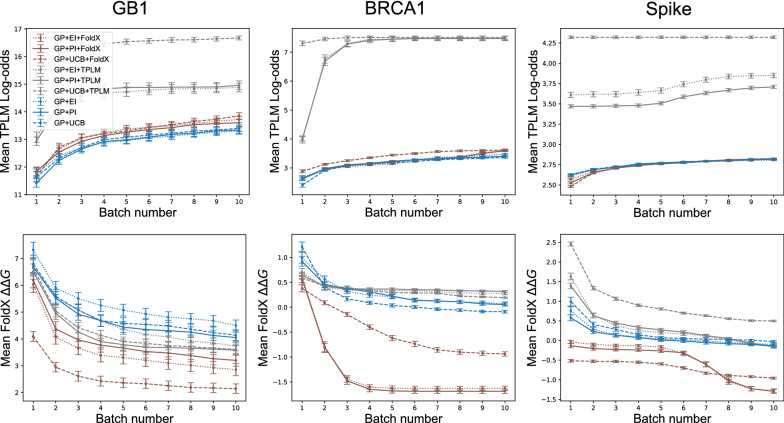
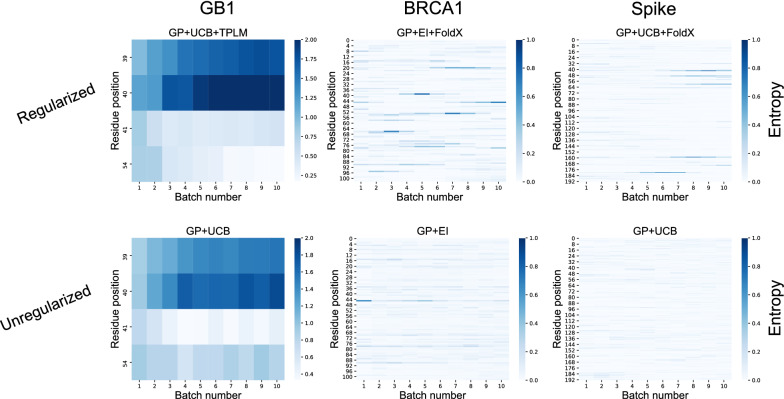
We applied our approach to DE to three representative proteins, GB1, BRCA1, and SARS-CoV-2 Spike, and evaluated both evolutionary and structure-based regularization terms. The results of these experiments demonstrate that: (i) structure-based regularization usually leads to better designs (and never hurts), compared to the unregularized setting; (ii) evolutionary-based regularization tends to be least effective; and (iii) regularization leads to better designs because it effectively focuses the search in certain areas of sequence space, making better use of the experimental budget. Additionally, like previous work in Machine learning assisted DE, we find that our approach significantly reduces the experimental burden of DE, relative to model-free methods.
Introducing regularization into a Bayesian ML-assisted DE framework alters the exploratory patterns of the underlying optimization routine, and can shift variant selections towards those with a range of targeted and desirable properties. In particular, we find that structure-based regularization often improves variant selection compared to unregularized approaches, and never hurts.
定向进化(DE)是一种蛋白质工程技术,它涉及多轮迭代诱变和筛选,以寻找能优化特定属性(如与指定靶标的结合亲和力)的序列。不幸的是,潜在的优化问题是欠定的,因此为改善指定属性而引入的突变可能会以未测量但同样重要的属性(如溶解度、热稳定性等)为代价。我们通过将DE表述为一个正则化贝叶斯优化问题来解决这个问题,其中正则化项反映了进化或基于结构的约束。
我们将我们的方法应用于DE,针对三种代表性蛋白质GB1、BRCA1和SARS-CoV-2刺突蛋白,并评估了基于进化和基于结构的正则化项。这些实验结果表明:(i)与未正则化的情况相比,基于结构的正则化通常会带来更好的设计(而且从不产生负面影响);(ii)基于进化的正则化往往效果最差;(iii)正则化能带来更好的设计,因为它有效地将搜索集中在序列空间的某些区域,从而更好地利用实验预算。此外,与机器学习辅助DE的先前工作一样,我们发现我们的方法相对于无模型方法显著减轻了DE的实验负担。
在贝叶斯机器学习辅助的DE框架中引入正则化会改变潜在优化程序的探索模式,并可将变体选择转向具有一系列目标和理想属性的变体。特别是,我们发现与未正则化的方法相比,基于结构的正则化通常会改善变体选择,而且从不产生负面影响。