Department of Computer Science, University of Helsinki, Helsinki 00014, Finland.
Bioinformatics. 2021 Dec 11;37(24):4611-4619. doi: 10.1093/bioinformatics/btab516.
Variant calling workflows that utilize a single reference sequence are the de facto standard elementary genomic analysis routine for resequencing projects. Various ways to enhance the reference with pangenomic information have been proposed, but scalability combined with seamless integration to existing workflows remains a challenge.
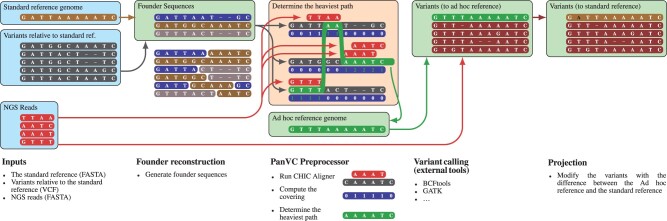
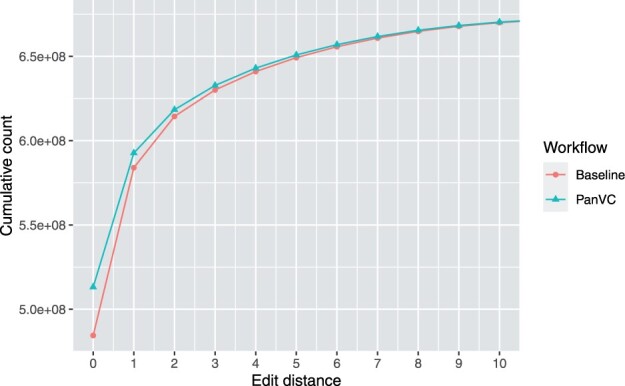
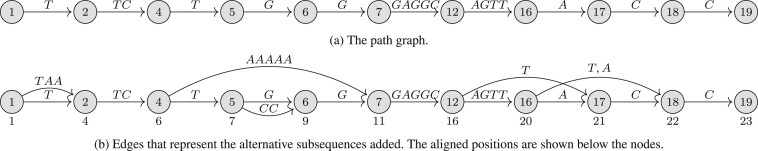
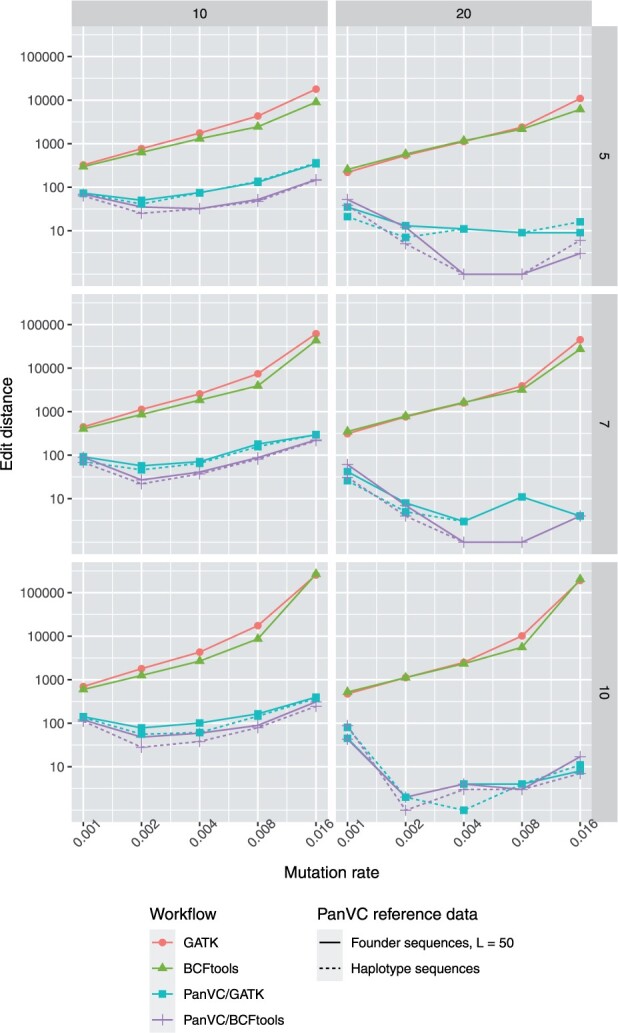
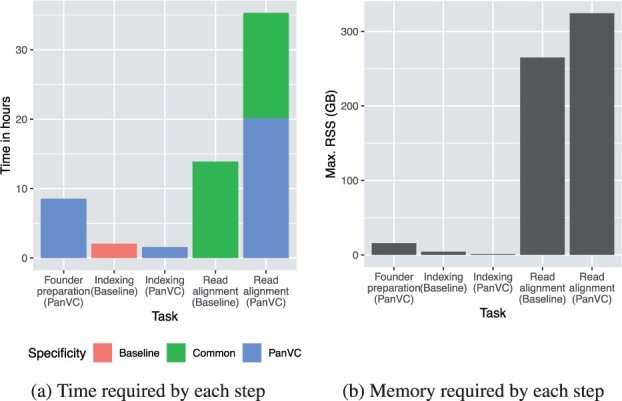
We present PanVC with founder sequences, a scalable and accurate variant calling workflow based on a multiple alignment of reference sequences. Scalability is achieved by removing duplicate parts up to a limit into a founder multiple alignment, that is then indexed using a hybrid scheme that exploits general purpose read aligners. Our implemented workflow uses GATK or BCFtools for variant calling, but the various steps of our workflow (e.g. vcf2multialign tool, founder reconstruction) can be of independent interest as a basis for creating novel pangenome analysis workflows beyond variant calling.
Our open access tools and instructions how to reproduce our experiments are available at the following address: https://github.com/algbio/panvc-founders.
Supplementary data are available at Bioinformatics online.
利用单一参考序列的变异调用工作流程是重测序项目基本的标准基因组分析常规。已经提出了各种利用泛基因组信息增强参考序列的方法,但可扩展性以及与现有工作流程的无缝集成仍然是一个挑战。
我们提出了 PanVC 与创始序列,这是一种基于参考序列多重比对的可扩展且准确的变异调用工作流程。可扩展性是通过将重复部分最多去除到创始多重比对的限制内来实现的,然后使用利用通用读取比对器的混合方案对其进行索引。我们实现的工作流程使用 GATK 或 BCFtools 进行变异调用,但我们工作流程的各个步骤(例如 vcf2multialign 工具、创始重建)可以作为创建超越变异调用的新型泛基因组分析工作流程的基础,具有独立的意义。
我们的开放访问工具以及重现我们实验的说明可在以下地址获得:https://github.com/algbio/panvc-founders。
补充数据可在生物信息学在线获得。