Serrano Jiménez Alfredo, Sánchez Muzas Alberto P, Zhang Yaolong, Ovčar Juraj, Jiang Bin, Lončarić Ivor, Juaristi J Iñaki, Alducin Maite
Centro de Física de Materiales CFM/MPC (CSIC-UPV/EHU), Paseo Manuel de Lardizabal 5, 20018 Donostia-San Sebastián, Spain.
Hefei National Laboratory for Physical Science at the Microscale, Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, Department of Chemical Physics, University of Science and Technology of China, Hefei, Anhui 230026, China.
J Chem Theory Comput. 2021 Aug 10;17(8):4648-4659. doi: 10.1021/acs.jctc.1c00347. Epub 2021 Jul 19.
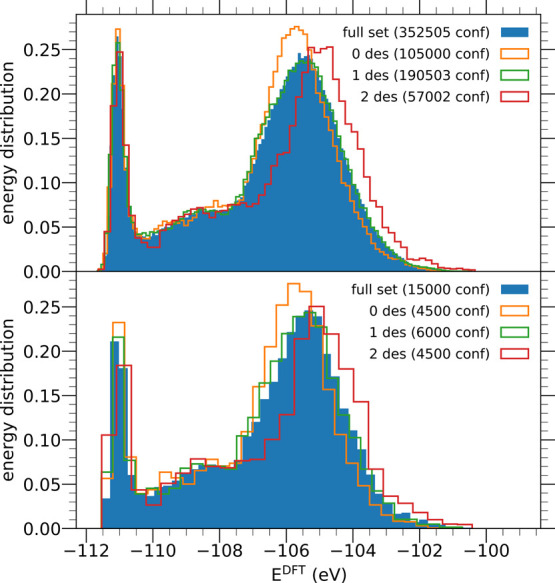
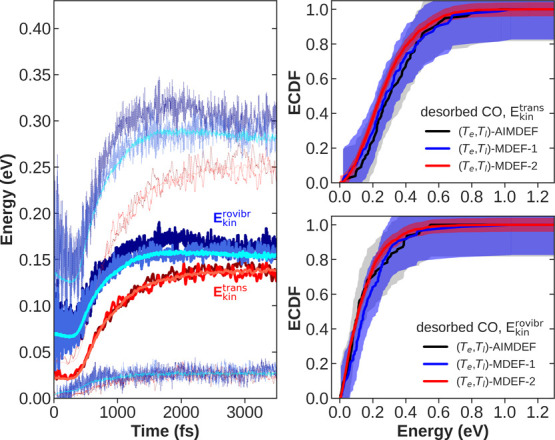
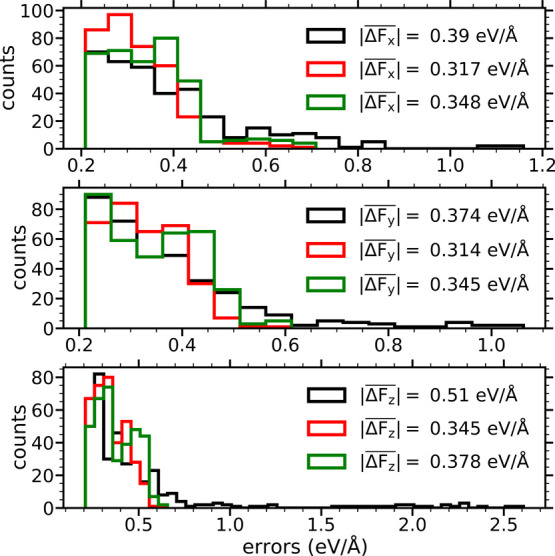
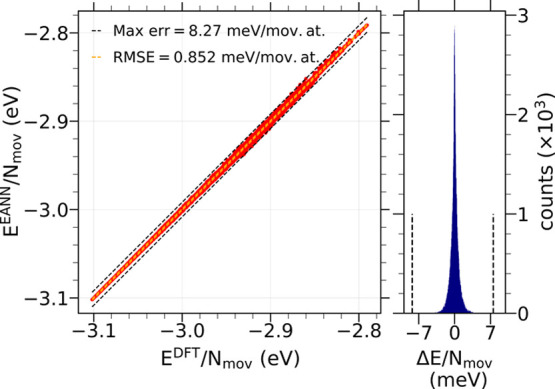
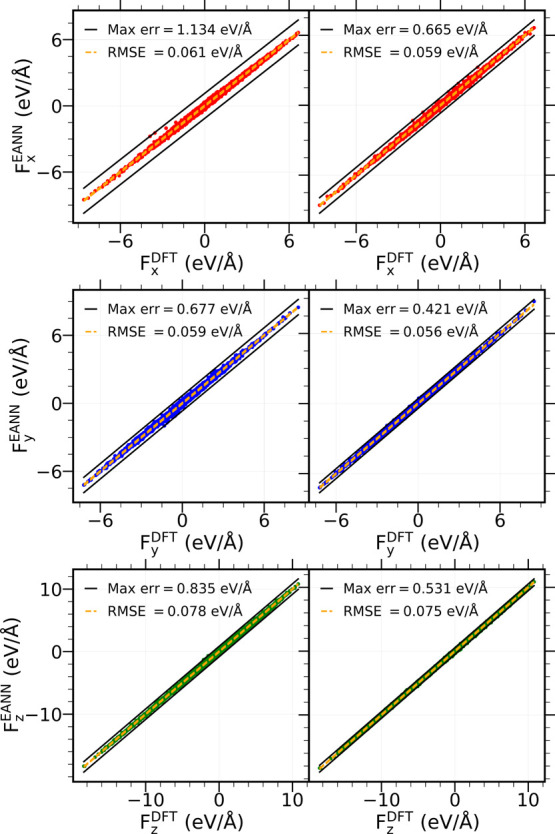
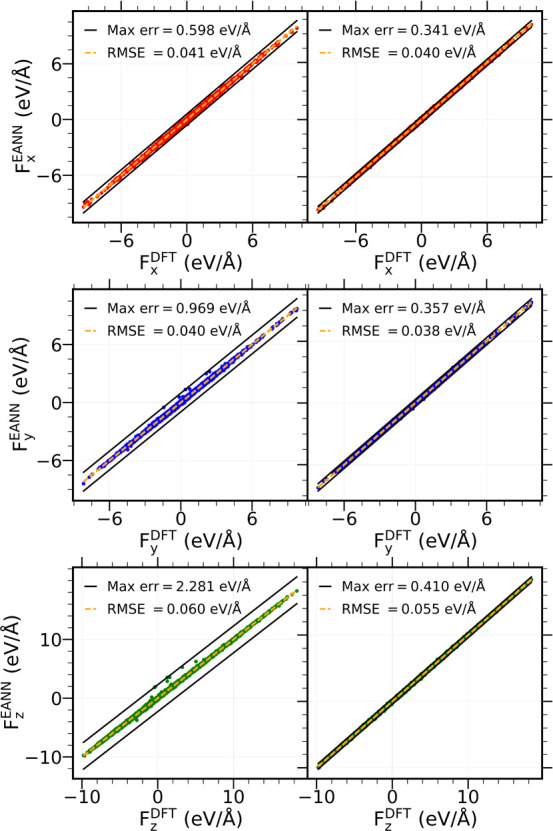
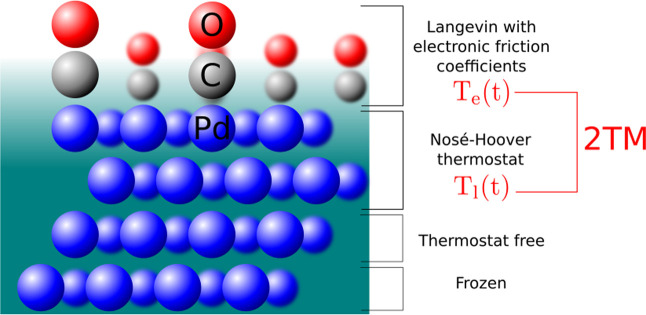
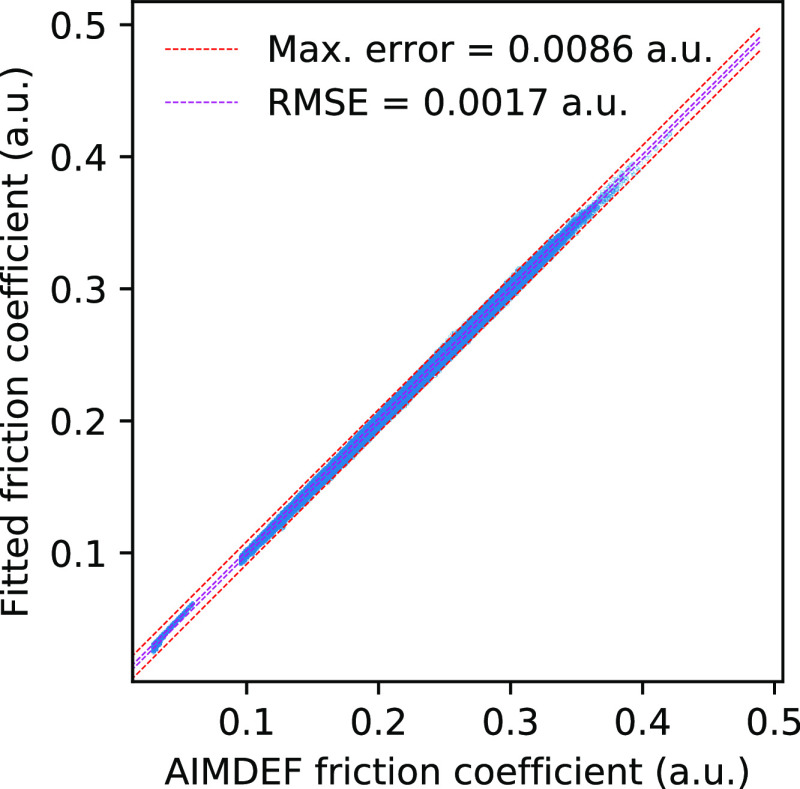
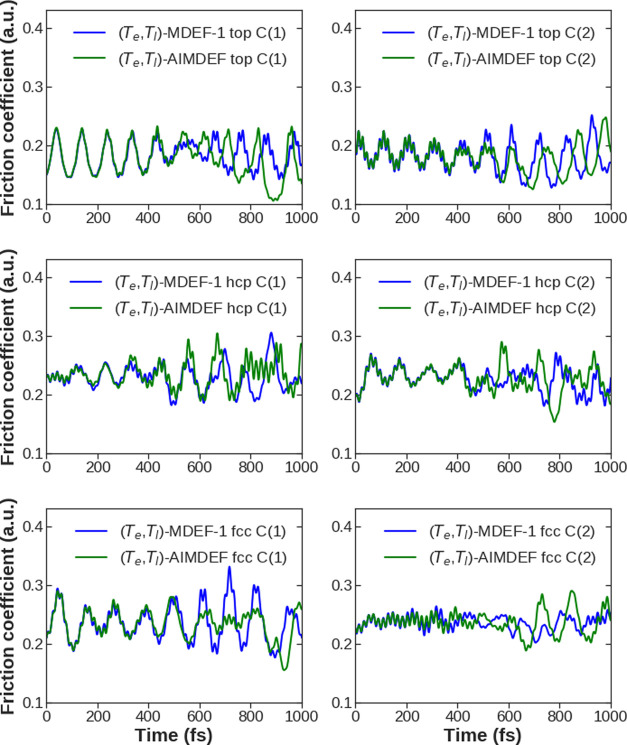
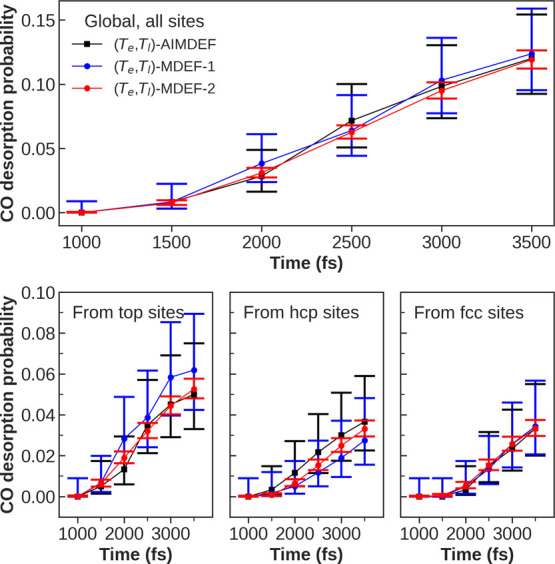
Modeling the ultrafast photoinduced dynamics and reactivity of adsorbates on metals requires including the effect of the laser-excited electrons and, in many cases, also the effect of the highly excited surface lattice. Although the recent ab initio molecular dynamics with electronic friction and thermostats, (,)-AIMDEF [Alducin, M.; 2019, 123, 246802], enables such complex modeling, its computational cost may limit its applicability. Here, we use the new embedded atom neural network (EANN) method [Zhang, Y.; 2019, 10, 4962] to develop an accurate and extremely complex potential energy surface (PES) that allows us a detailed and reliable description of the photoinduced desorption of CO from the Pd(111) surface with a coverage of 0.75 monolayer. Molecular dynamics simulations performed on this EANN-PES reproduce the (,)-AIMDEF results with a remarkable level of accuracy. This demonstrates the outstanding performance of the obtained EANN-PES that is able to reproduce available density functional theory (DFT) data for an extensive range of surface temperatures (90-1000 K); a large number of degrees of freedom, those corresponding to six CO adsorbates and 24 moving surface atoms; and the varying CO coverage caused by the abundant desorption events.
对金属表面吸附物的超快光致动力学和反应性进行建模,需要考虑激光激发电子的影响,并且在许多情况下,还需要考虑高激发表面晶格的影响。尽管最近采用带有电子摩擦和恒温器的从头算分子动力学方法((,)-AIMDEF [阿尔杜辛,M.;2019年,123卷,246802页])能够进行这种复杂的建模,但其计算成本可能会限制其适用性。在这里,我们使用新的嵌入原子神经网络(EANN)方法[张,Y.;2019年,10卷,4962页]来开发一个精确且极其复杂的势能面(PES),该势能面使我们能够详细且可靠地描述覆盖度为0.75单层的CO从Pd(111)表面的光致脱附。在这个EANN-PES上进行的分子动力学模拟以极高的精度重现了(,)-AIMDEF的结果。这证明了所获得的EANN-PES的卓越性能,它能够在广泛的表面温度范围(90 - 1000 K)内重现可用的密度泛函理论(DFT)数据;对应于六个CO吸附物和24个移动表面原子的大量自由度;以及由大量脱附事件引起的变化的CO覆盖度。