Nickoloff Jac A, Sharma Neelam, Allen Christopher P, Taylor Lynn, Allen Sage J, Jaiswal Aruna S, Hromas Robert
Department of Environmental and Radiological Health Sciences, Colorado State University, Fort Collins, CO, USA.
Department of Microbiology, Immunology and Pathology, Flow Cytometry and Cell Sorting Facility, Colorado State University, Fort Collins, CO, USA.
Int J Radiat Biol. 2023;99(6):903-914. doi: 10.1080/09553002.2021.1956001. Epub 2021 Aug 4.
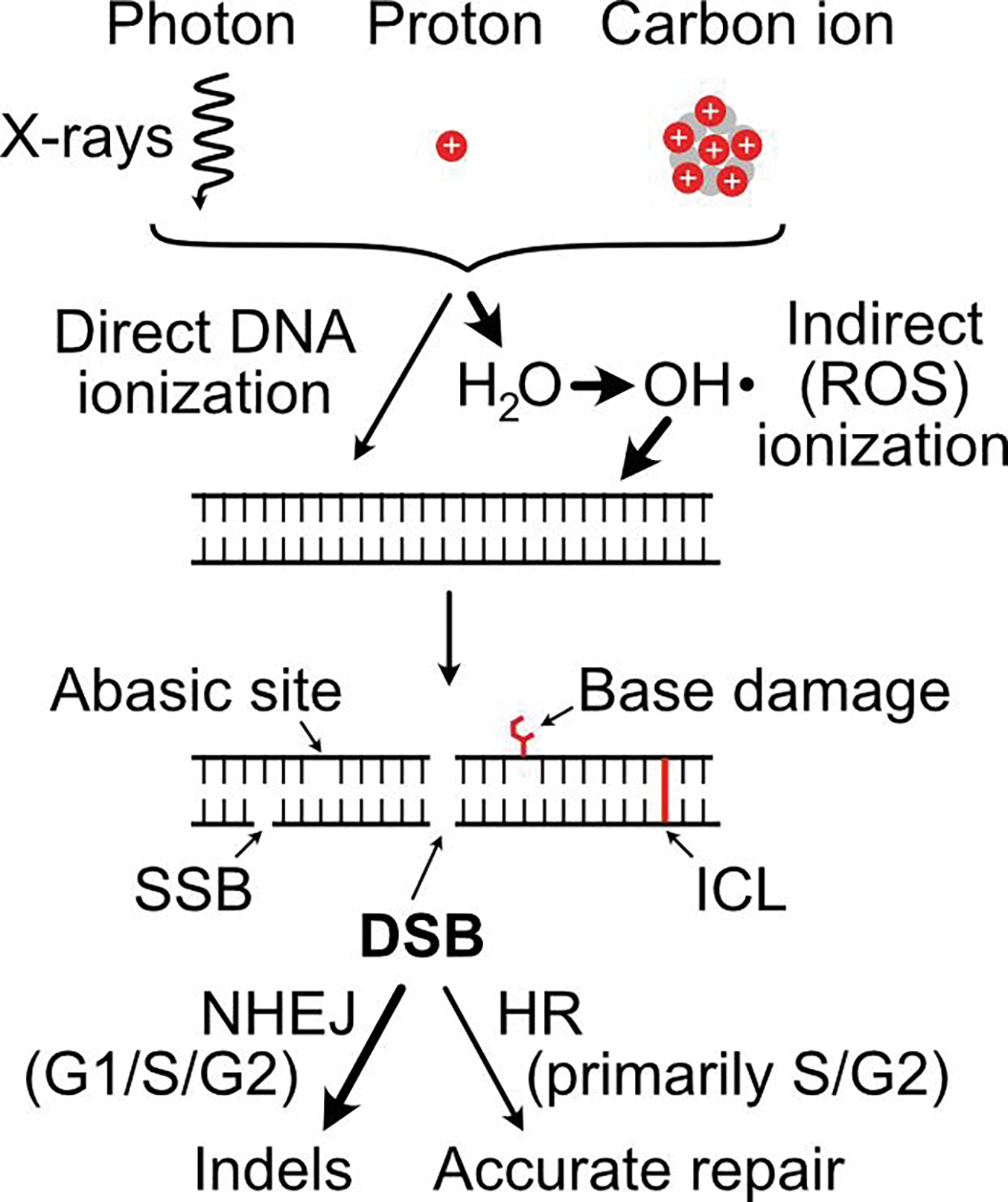
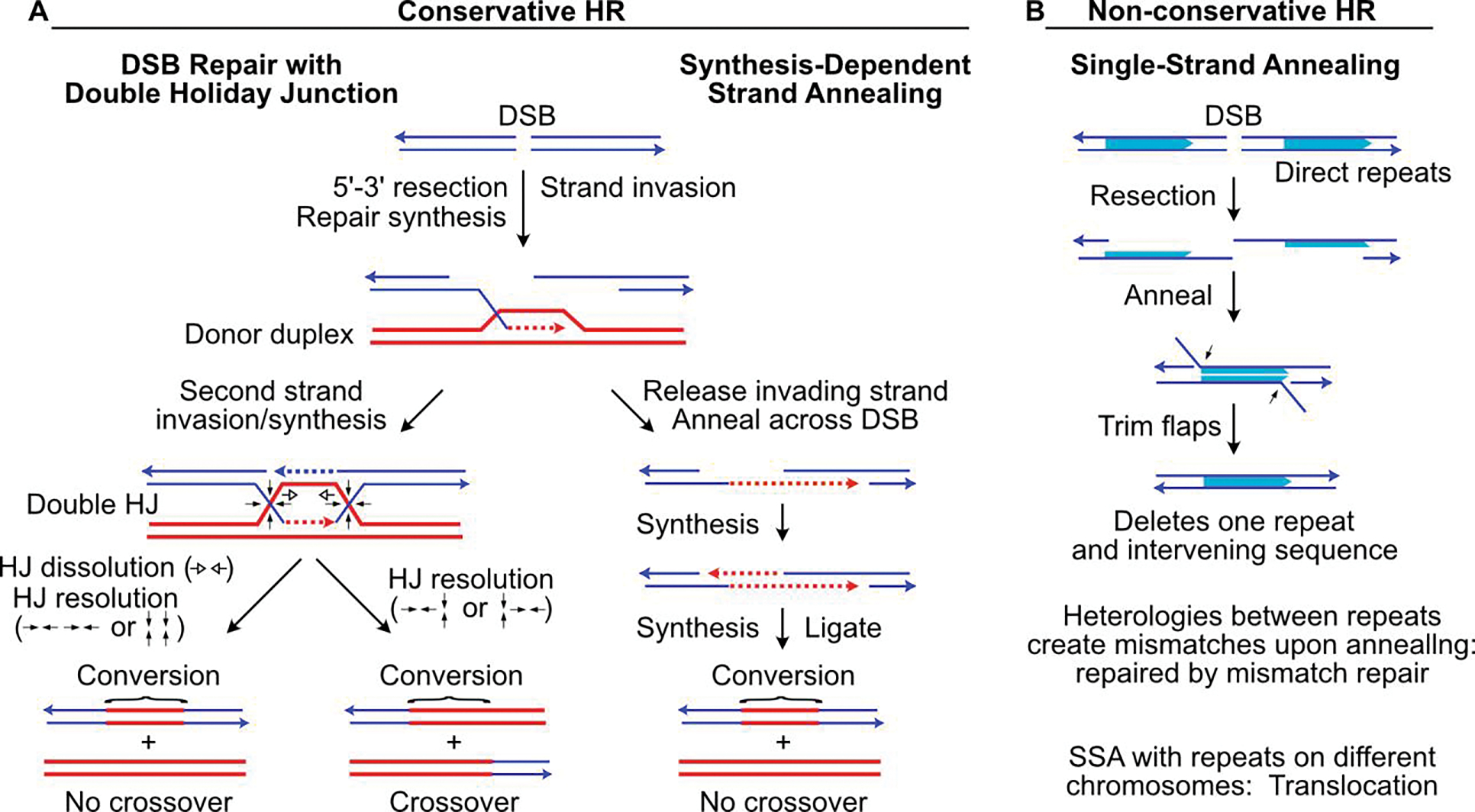
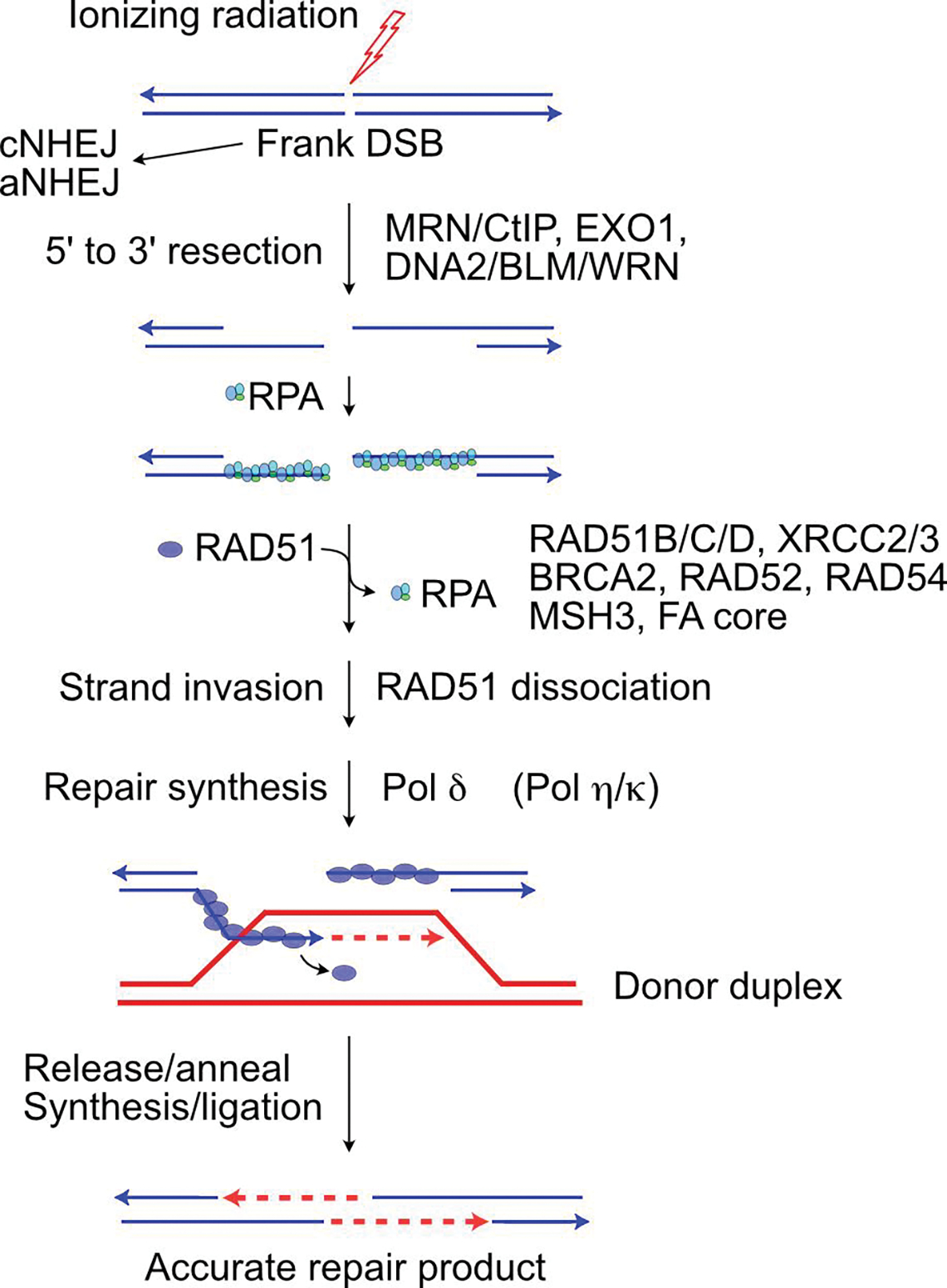
Ionizing radiation induces a vast array of DNA lesions including base damage, and single- and double-strand breaks (SSB, DSB). DSBs are among the most cytotoxic lesions, and mis-repair causes small- and large-scale genome alterations that can contribute to carcinogenesis. Indeed, ionizing radiation is a 'complete' carcinogen. DSBs arise immediately after irradiation, termed 'frank DSBs,' as well as several hours later in a replication-dependent manner, termed 'secondary' or 'replication-dependent DSBs. DSBs resulting from replication fork collapse are single-ended and thus pose a distinct problem from two-ended, frank DSBs. DSBs are repaired by error-prone nonhomologous end-joining (NHEJ), or generally error-free homologous recombination (HR), each with sub-pathways. Clarifying how these pathways operate in normal and tumor cells is critical to increasing tumor control and minimizing side effects during radiotherapy.
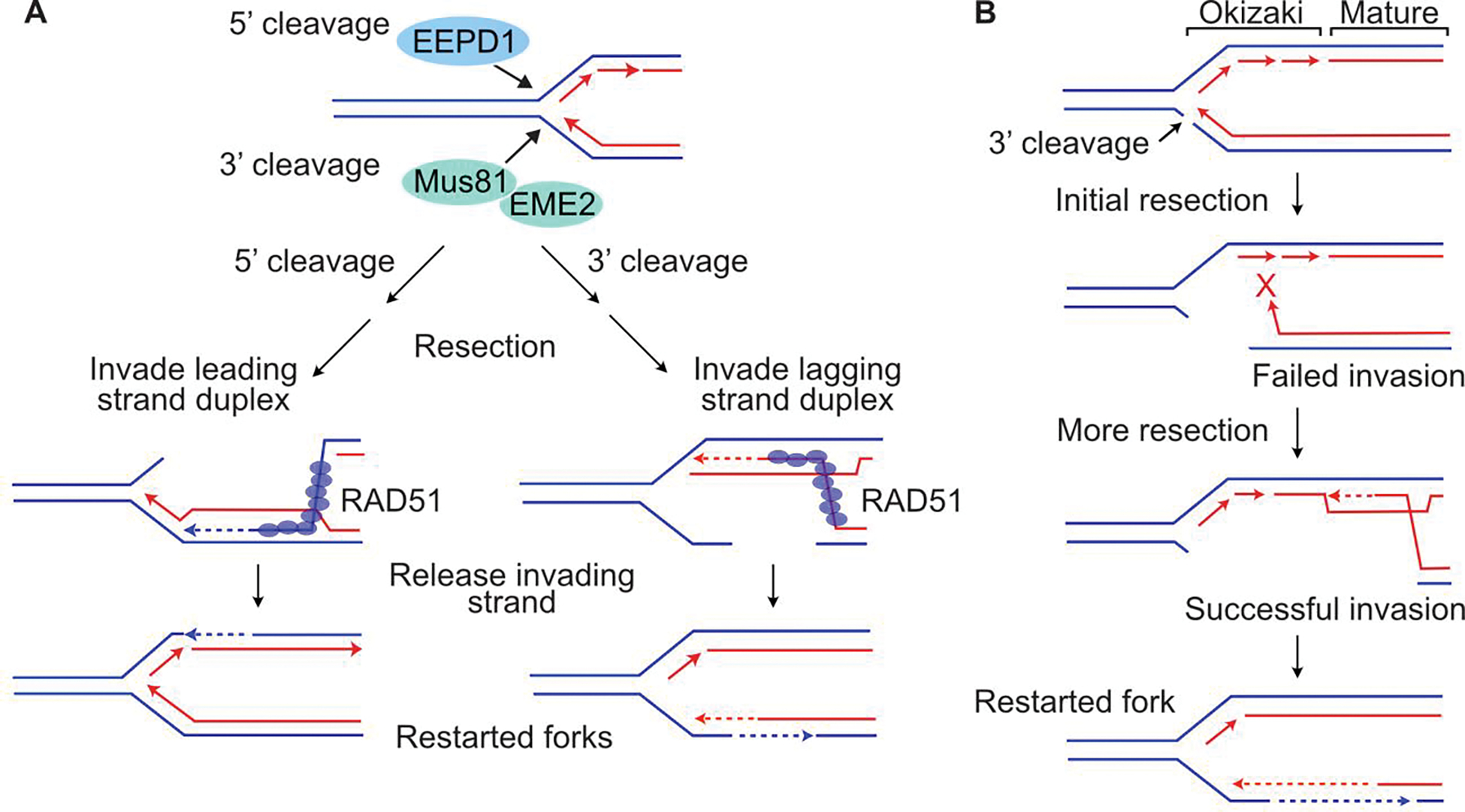
The choice between NHEJ and HR is regulated during the cell cycle and by other factors. DSB repair pathways are major contributors to cell survival after ionizing radiation, including tumor-resistance to radiotherapy. Several nucleases are important for HR-mediated repair of replication-dependent DSBs and thus replication fork restart. These include three structure-specific nucleases, the 3' MUS81 nuclease, and two 5' nucleases, EEPD1 and Metnase, as well as three end-resection nucleases, MRE11, EXO1, and DNA2. The three structure-specific nucleases evolved at very different times, suggesting incremental acceleration of replication fork restart to limit toxic HR intermediates and genome instability as genomes increased in size during evolution, including the gain of large numbers of HR-prone repetitive elements. Ionizing radiation also induces delayed effects, observed days to weeks after exposure, including delayed cell death and delayed HR. In this review we highlight the roles of HR in cellular responses to ionizing radiation, and discuss the importance of HR as an exploitable target for cancer radiotherapy.
电离辐射可诱发大量DNA损伤,包括碱基损伤、单链和双链断裂(SSB、DSB)。双链断裂是细胞毒性最强的损伤之一,错误修复会导致小规模和大规模的基因组改变,进而可能引发癌症。事实上,电离辐射是一种“完全”致癌物。双链断裂在照射后立即出现,称为“明显双链断裂”,也会在数小时后以复制依赖的方式出现,称为“继发性”或“复制依赖型双链断裂”。复制叉坍塌导致的双链断裂是单端的,因此与两端的明显双链断裂构成不同的问题。双链断裂通过易出错的非同源末端连接(NHEJ)或通常无错误的同源重组(HR)进行修复,每种方式都有子途径。阐明这些途径在正常细胞和肿瘤细胞中的运作方式对于提高肿瘤控制率和在放疗期间将副作用降至最低至关重要。
非同源末端连接和同源重组之间的选择在细胞周期及其他因素的调控下进行。双链断裂修复途径是电离辐射后细胞存活的主要因素,包括肿瘤对放疗的抗性。几种核酸酶对于同源重组介导的复制依赖型双链断裂修复以及复制叉重启很重要。这些核酸酶包括三种结构特异性核酸酶、3'端MUS81核酸酶以及两种5'端核酸酶EEPD1和甲基转移酶,还有三种末端切除核酸酶MRE11、EXO1和DNA2。这三种结构特异性核酸酶在非常不同的时期进化,这表明随着进化过程中基因组大小的增加,包括大量易于发生同源重组的重复元件的获得,复制叉重启逐渐加速,以限制有毒的同源重组中间体和基因组不稳定性。电离辐射还会诱发延迟效应,在暴露后数天至数周观察到,包括延迟细胞死亡和延迟同源重组。在本综述中,我们强调了同源重组在细胞对电离辐射反应中的作用,并讨论了同源重组作为癌症放疗可利用靶点的重要性。